ADAR
Topic: Biology
 From HandWiki - Reading time: 15 min
From HandWiki - Reading time: 15 min
 Generic protein structure example |
The double-stranded RNA-specific adenosine deaminase enzyme family are encoded by the ADAR family genes.[1] ADAR stands for adenosine deaminase acting on RNA.[2][3] This article focuses on the ADAR proteins; This article details the evolutionary history, structure, function, mechanisms and importance of all proteins within this family.[1]
ADAR enzymes bind to double-stranded RNA (dsRNA) and convert adenosine to inosine (hypoxanthine) by deamination.[4] ADAR proteins act post-transcriptionally, changing the nucleotide content of RNA.[5] The conversion from adenosine to inosine (A to I) in the RNA disrupts the normal A:U pairing, destabilizing the RNA. Inosine is structurally similar to guanine (G) which leads to inosine to cytosine (I:C) binding.[6] Inosine typically mimics guanosine during translation but can also bind to uracil, cytosine, and adenosine, though it is not favored.
Codon changes may arise from RNA editing leading to changes in the coding sequences for proteins and their functions.[7] Most editing sites are found in noncoding regions of RNA such as untranslated regions (UTRs), Alu elements, and long interspersed nuclear elements (LINEs).[8] Codon changes can give rise to alternate transcriptional splice variants. ADAR impacts the transcriptome in editing-independent ways, likely by interfering with other RNA-binding proteins.[5]
Dysregulation of ADAR is associated with several diseases. Recent research supports a linkage between RNA-editing and nervous system disorders such as amyotrophic lateral sclerosis (ALS). Atypical RNA editing linked to ADAR may also correlate to mental disorders such as schizophrenia, epilepsy, and suicidal depression.[9]
Discovery
The ADAR enzyme and its associated gene were discovered accidentally in 1987 as a result of research by Brenda Bass and Harold Weintraub.[10] These researchers were using antisense RNA inhibition to determine which genes play a key role in the development of Xenopus laevis embryos. Previous research on Xenopus oocytes was successful. However, when Bass and Weintraub applied identical protocols to Xenopus embryos, they were unable to determine the embryo's developmental genes. To understand why the method was unsuccessful, they began comparing duplex RNA in both oocytes and embryos. This led them to discover a developmentally regulated activity that denatures RNA:RNA hybrids in embryos.
In 1988, Richard Wagner et al. further studied the activity occurring on Xenopus embryos.[11] They determined a protein was responsible for unwinding of RNA due to the absence of activity after proteinase treatment. This protein is specific for dsRNA and does not require ATP. It became evident this protein's activity on dsRNA modifies it beyond a point of rehybridization but does not fully denature it. Finally, the researchers determined this unwinding is due to the deamination of adenosine residues to inosine. This modification results in mismatched base-pairing between inosine and uridine, leading to the destabilization and unwinding of dsRNA.
Evolution and function
ADARs are one of the most common forms of RNA editing, and have both selective and non-selective activity.[12] ADAR is able to modify and regulate the output of gene product, as inosine is interpreted by the cell to be guanosine. ADAR can change the functionality of small RNA molecules. Recently, ADARs have also been discovered as a regulator on splicing and circRNA biogenesis with their editing capability or RNA binding function.[13][14][15] It is believed that ADAR evolved from ADAT (Adenosine Deaminase Acting on tRNA), a critical protein present in all eukaryotes, early in the metazoan period through the addition of a dsRNA binding domain. This likely occurred in the lineage which leads to the crown Metazoa. When a duplicate ADAT gene was coupled to another gene which encoded at least one double stranded RNA binding. The ADAR family of genes has been largely conserved over the history of its existence. This, along with its presence in the majority of modern phyla, indicates that RNA editing is essential in regulating genes for metazoan organisms. ADAR has not been discovered in a variety of non-metazoan eukaryotes, such as plants, fungi and choanoflagellates.
ADARs are suggested to have two functions: to increase diversity of the proteome by inducing creation of harmless non-genomically encoded proteins, and protecting crucial translational sites. The conventional belief is their primary role is to increase the diversity of transcripts and expand the protein variation, promoting evolution of proteins.[1]
Forms of ADAR enzymes
In mammals, there are three types of ADAR enzymes: ADAR (ADAR1), ADARB1 (ADAR2), and ADARB2 (ADAR3).[1]
ADAR (ADAR1) and ADAR2 (ADARB1)
ADAR one and two are both found within various tissues of the body. These two forms of ADAR are also found to be catalytically active, meaning they can be used as a catalyst in a reaction. Both forms also have similar expression pattern structures of proteins and require substrate double-stranded RNA structures.[7] However, they differ in their editing activity in that both ADAR one and two can edit GluR-B pre-mRNA at the R/G site and only ADAR2 can alter the Q/R site.[16] ADAR1 has been found two have two isoforms, ADAR1p150 and ADARp110. ADAR1p110 is typically found in the nucleus, while ADAR1p150 shuffles between the nucleus and the cytoplasm, mostly present in the cytoplasm.
ADAR3 (ADARB2)
ADAR 3 varies from the other two forms of ADAR in that it is only found within brain tissue. It also is considered to be inactive when it comes to catalytic activity.[7] ADAR3 has been found to be linked to memory and learning in mice, showing that it plays a crucial role in the nervous system. In vitro studies have also shown that ADAR3 might play a role in the regulation of ADAR one and two.[17]
Catalytic activity
Biochemical reaction
ADARs catalyze the hydrolytic deamination reaction from adenosine to inosine.[4] An activated water molecule will react with adenosine in a nucleophilic substitution reaction with the carbon-6 amine group. A hydrated intermediate will exist for a short period of time, then the amine group will leave as an ammonia ion.
-
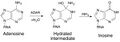 Adenosine conversion to Inosine via ADAR
Adenosine conversion to Inosine via ADAR
Active site
In humans, ADAR enzymes have two to three amino-terminal dsRNA binding domains (dsRBDs), and one carboxy terminal catalytic deaminase domain.[18] In the dsRBD there is a conserved α-β-β-β-α configuration.[7] ADAR1 contains two areas for binding Z-DNA known as Zα and Zβ.[19][20] ADAR2 and ADAR3 have an arginine rich single stranded RNA (ssRNA) binding domain. A crystal structure of ADAR2 has been solved.[18] In the enzyme active site, there is a glutamic acid residue(E396) that hydrogen bonds to a water. A histidine (H394) and two cysteine residues (C451 and C516) coordinate with a zinc ion. The zinc activates the water molecule for the nucleophilic hydrolytic deamination. Within the catalytic core there is an inositol hexakisphosphate (IP6), which stabilizes arginine and lysine residues.

Dimerization
ADAR1 and ADAR2 have been shown to form homodimers in mammals, while ADAR3 does not.[7] Earlier studies suggested that dimerization might be necessary for enzymatic activity and could occur independently of RNA binding, based on experiments with ADAR mutants that failed to bind double-stranded RNA (dsRNA) but could still dimerize, indicating protein-protein interactions were sufficient.[7][21] However, more recent research has clarified that dimerization is not strictly required for ADAR1 enzymatic activity. A recent study demonstrated that ADAR1 dimerization occurs specifically through its third double-stranded RNA-binding domain (dsRBD3), and importantly, this dimerization is RNA-independent.[22] This shows that ADAR1 can form dimers via a defined protein-protein interface without involving RNA. In addition, dimerization disruption do not completely abrogate ADAR1 editing activity, but affects editing efficiency differently depending on the selected editing site.[22]
Role in disease
Aicardi–Goutières syndrome and bilateral striatal necrosis/dystonia
ADAR1 is one of multiple genes which often contribute to Aicardi–Goutières syndrome when mutated.[23] Aicardi–Goutières syndrome is a genetic inflammatory disease primarily affecting the skin and the brain and it is characterized by high levels of IFN-α in cerebral spinal fluid.[24] The inflammation is caused by incorrect activation of interferon inducible genes such as those activated to fight off viral infections. Mutation and loss of function of ADAR1 prevents destabilization of double stranded RNA (dsRNA).[25] This buildup of dsRNA stimulates IFN production without a viral infection, causing an inflammatory reaction and autoimmune response.[26] The phenotype in the knock-out mice is rescued by the p150 form of ADAR1 containing the Zα domain that binds specifically to the left-handed double-stranded conformation found in Z-DNA and Z-RNA, but not by the p110 isoform lacking this domain.[27] In humans, the P193A mutation in the Zα domain is causal for Aicardi–Goutières syndrome[23] and for the more severe phenotype found in Bilateral Striatal Necrosis/Dystonia.[28] The findings establish a biological role for the left-handed Z-DNA conformation.[29]
Amyotrophic Lateral Sclerosis (ALS)
In motor neurons, the most well-grounded marker of amyotrophic lateral sclerosis (ALS) is the TAR DNA-binding protein (TDP-43). When there is failure of RNA-editing due to downregulation of TDP-43, motor neurons devoid of ADAR2 enzymes express unregulated, leading to abnormally permeable Ca2+ channels. ADAR2 knockout mice show signs of ALS phenotype similarity. Current researchers are developing a molecular targeting therapy by normalizing expression of ADAR2.[30]
Cancer
(ADAR)-induced A-to-I RNA editing may elicit dangerous amino acid mutations. Editing mRNA typically imparts missense mutations leading to alterations in the beginning and terminating regions of translation. However, crucial amino acid changes can occur, resulting in change of function of several cellular processes. Amino acid changes can result in protein structural changes at secondary, tertiary, and quaternary structures. Researchers observed high levels of oncogenetic A-to-I editing in circular RNA precursors, directly confirming ADAR's relationship to cancer.[15] A list of tumor related RNA editing sites can be found here.[31]
Hepatocellular carcinoma
Studies of patients with hepatocellular carcinoma (HCC) have shown trends of upregulated ADAR1 and downregulated ADAR2. Results suggest the irregular regulation is responsible for the disrupted A to I editing pattern seen in HCC and that ADAR1 acts as an oncogene in this context whilst ADAR2 has tumor suppressor activities.[32] The imbalance in ADAR expression could change the frequency of A to I transitions in the protein coding region of genes, resulting in mutated proteins which drive the disease. The dysregulation of ADAR1 and ADAR2 could be used as a possible prognostic marker.
Melanoma
Studies have indicated that loss of ADAR1 contributes to melanoma growth and metastasis. ADAR enzymes can act on microRNA and affect its biogenesis, stability and/or its binding target.[33] ADAR1 may be downregulated by cAMP- response element binding protein (CREB), limiting its ability to act on miRNA.[34] One such example is miR-455-5p which is edited by ADAR1. When ADAR is downregulated by CREB the unedited miR-455-5p downregulates a tumor suppressor protein called CPEB1, contributing to melanoma progression in an in vivo model.
Dyschromatosis symmetrica hereditaria (DSH1)
A Gly1007Arg mutation in ADAR1, as well as other truncated versions, have been implicated as a cause in some cases of DSH1.[35] This is a disease characterized by hyperpigmentation in the hands and feet and can occur in Japanese and Chinese families.
HIV
Expression levels of the ADAR1 protein have shown to be elevated during HIV infection and it has been suggested that it is responsible for A to G mutations in the HIV genome, inhibiting replication.[36] The mutation in the HIV genome by ADAR1 might in some cases lead to beneficial viral mutations which could contribute to drug resistance.
Viral activity
Antiviral
ADAR1 is an interferon ( IFN )-inducible protein (one released by a cell in response to a pathogen or virus), able to assist in a cell's immune pathway. Evidence shows elimination of HCV replicon, Lymphocytic choriomeningitis LCMV, and polyomavirus.[37][38]
Proviral
ADAR1 is proviral in other circumstances. ADAR1's A to I editing has been found in many viruses including measles virus,[39][38][40] influenza virus,[41] lymphocytic choriomeningitis virus,[42] polyomavirus,[43] hepatitis delta virus,[44] and hepatitis C virus.[45] Although ADAR1 has been seen in other viruses, it has only been studied extensively in a few. Research on measles virus shows ADAR1 enhancing viral replication through two different mechanisms: RNA editing and inhibition of dsRNA-activated protein kinase (PKR).[37][38] Specifically, viruses are thought to use ADAR1 as a positive replication factor by selectively suppressing dsRNA-dependent and antiviral pathways.[46]
See also
- RNA editing
- Potassium channel RNA editing signal
- ADARB1
- Z-DNA
References
- ↑ 1.0 1.1 1.2 1.3 "The ADAR protein family". Genome Biology 13 (12): 252. December 2012. doi:10.1186/gb-2012-13-12-252. PMID 23273215.
- ↑ "Entrez Gene: ADAR Adenosine Deaminase Acting on RNA". https://www.ncbi.nlm.nih.gov/gene?Db=gene&Cmd=ShowDetailView&TermToSearch=103.
- ↑ "Molecular cloning of cDNA for double-stranded RNA adenosine deaminase, a candidate enzyme for nuclear RNA editing". Proceedings of the National Academy of Sciences of the United States of America 91 (24): 11457–11461. November 1994. doi:10.1073/pnas.91.24.11457. PMID 7972084. Bibcode: 1994PNAS...9111457K.
- ↑ 4.0 4.1 Adenosine deaminases acting on RNA (ADARs) and A-to-I editing. Heidelberg: Springer. 2012. ISBN 978-3-642-22800-1.
- ↑ 5.0 5.1 "ADAR". U.S. National Library of Medicine. https://www.ncbi.nlm.nih.gov/gene/10.
- ↑ "Inosine induces context-dependent recoding and translational stalling". Nucleic Acids Research 47 (1): 3–14. January 2019. doi:10.1093/nar/gky1163. PMID 30462291.
- ↑ 7.0 7.1 7.2 7.3 7.4 7.5 "Functions and regulation of RNA editing by ADAR deaminases". Annual Review of Biochemistry 79 (1): 321–349. 7 June 2010. doi:10.1146/annurev-biochem-060208-105251. PMID 20192758. Bibcode: 2010ARBio..79..321N.
- ↑ "The dynamic epitranscriptome: A to I editing modulates genetic information". Chromosoma 125 (1): 51–63. March 2016. doi:10.1007/s00412-015-0526-9. PMID 26148686.
- ↑ "The ADAR protein family". Genome Biology 13 (12): 252. December 2012. doi:10.1186/gb-2012-13-12-252. PMID 23273215.
- ↑ "Adenosine deaminases acting on RNA (ADARs) are both antiviral and proviral". Virology 411 (2): 180–193. March 2011. doi:10.1016/j.virol.2010.12.004. PMID 21211811.
- ↑ "A double-stranded RNA unwinding activity introduces structural alterations by means of adenosine to inosine conversions in mammalian cells and Xenopus eggs". Proceedings of the National Academy of Sciences of the United States of America 86 (8): 2647–2651. April 1989. doi:10.1073/pnas.86.8.2647. PMID 2704740. Bibcode: 1989PNAS...86.2647W.
- ↑ "The origin of the ADAR gene family and animal RNA editing". BMC Evolutionary Biology 15 (1). January 2015. doi:10.1186/s12862-015-0279-3. PMID 25630791. Bibcode: 2015BMCEE..15....4G.
- ↑ "Cis- and trans-regulations of pre-mRNA splicing by RNA editing enzymes influence cancer development". Nature Communications 11 (1). February 2020. doi:10.1038/s41467-020-14621-5. PMID 32034135. Bibcode: 2020NatCo..11..799T.
- ↑ "RNA editing in nascent RNA affects pre-mRNA splicing". Genome Research 28 (6): 812–823. June 2018. doi:10.1101/gr.231209.117. PMID 29724793.
- ↑ 15.0 15.1 "ADARs act as potent regulators of circular transcriptome in cancer". Nature Communications 13 (1). March 2022. doi:10.1038/s41467-022-29138-2. PMID 35314703. Bibcode: 2022NatCo..13.1508S.
- ↑ Källman, Annika M.; Sahlin, Margareta; Ohman, Marie (2003-08-15). "ADAR2 A-->I editing: site selectivity and editing efficiency are separate events". Nucleic Acids Research 31 (16): 4874–4881. doi:10.1093/nar/gkg681. ISSN 1362-4962. PMID 12907730.
- ↑ Wang, Yuru; Chung, Dong Hee; Monteleone, Leanna R.; Li, Jie; Chiang, Yao; Toney, Michael D.; Beal, Peter A. (2019-11-18). "RNA binding candidates for human ADAR3 from substrates of a gain of function mutant expressed in neuronal cells". Nucleic Acids Research 47 (20): 10801–10814. doi:10.1093/nar/gkz815. ISSN 1362-4962. PMID 31552420.
- ↑ 18.0 18.1 "The ADAR protein family". Genome Biology 13 (12): 252. December 2012. doi:10.1186/gb-2012-13-12-252. PMID 23273215.
- ↑ "Thermodynamic analysis of Zα domain-nucleic acid interactions". The Biochemical Journal 479 (16): 1727–1741. August 2022. doi:10.1042/BCJ20220200. PMID 35969150.
- ↑ "Enrichment of Zα domains at cytoplasmic stress granules is due to their innate ability to bind to nucleic acids". Journal of Cell Science 134 (10). May 2021. doi:10.1242/jcs.258446. PMID 34037233.
- ↑ "Requirement of dimerization for RNA editing activity of adenosine deaminases acting on RNA". The Journal of Biological Chemistry 278 (19): 17093–17102. May 2003. doi:10.1074/jbc.M213127200. PMID 12618436.
- ↑ 22.0 22.1 "Dimerization of ADAR1 modulates site-specificity of RNA editing". Nature Communications 15 (1). November 2024. doi:10.1038/s41467-024-53777-2. PMID 39572551. Bibcode: 2024NatCo..1510051M.
- ↑ 23.0 23.1 "Mutations in ADAR1 cause Aicardi-Goutières syndrome associated with a type I interferon signature". Nature Genetics 44 (11): 1243–1248. November 2012. doi:10.1038/ng.2414. PMID 23001123.
- ↑ "Adenosine deaminase acting on RNA 1 limits RIG-I RNA detection and suppresses IFN production responding to viral and endogenous RNAs". Journal of Immunology 193 (7): 3436–3445. October 2014. doi:10.4049/jimmunol.1401136. PMID 25172485.
- ↑ "ADAR RNA editing in human disease; more to it than meets the I". Human Genetics 136 (9): 1265–1278. September 2017. doi:10.1007/s00439-017-1837-0. PMID 28913566.
- ↑ "RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself". Science 349 (6252): 1115–1120. September 2015. doi:10.1126/science.aac7049. PMID 26275108. Bibcode: 2015Sci...349.1115L.
- ↑ "RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis". Proceedings of the National Academy of Sciences of the United States of America 108 (1): 331–336. January 2011. doi:10.1073/pnas.1017241108. PMID 21173229. Bibcode: 2011PNAS..108..331W.
- ↑ "A type I interferon signature identifies bilateral striatal necrosis due to mutations in ADAR1". Journal of Medical Genetics 51 (2): 76–82. February 2014. doi:10.1136/jmedgenet-2013-102038. PMID 24262145.
- ↑ "Mendelian disease caused by variants affecting recognition of Z-DNA and Z-RNA by the Zα domain of the double-stranded RNA editing enzyme ADAR". European Journal of Human Genetics 28 (1): 114–117. January 2020. doi:10.1038/s41431-019-0458-6. PMID 31320745.
- ↑ "Cell death cascade and molecular therapy in ADAR2-deficient motor neurons of ALS". Neuroscience Research 144: 4–13. July 2019. doi:10.1016/j.neures.2018.06.004. PMID 29944911.
- ↑ "A-to-I RNA Editing in Cancer: From Evaluating the Editing Level to Exploring the Editing Effects". Frontiers in Oncology 10. 2021. doi:10.3389/fonc.2020.632187. PMID 33643923.
- ↑ "A disrupted RNA editing balance mediated by ADARs (Adenosine DeAminases that act on RNA) in human hepatocellular carcinoma". Gut 63 (5): 832–843. May 2014. doi:10.1136/gutjnl-2012-304037. PMID 23766440.
- ↑ "Editing independent effects of ADARs on the miRNA/siRNA pathways". The EMBO Journal 28 (20): 3145–3156. October 2009. doi:10.1038/emboj.2009.244. PMID 19713932.
- ↑ 34.0 34.1 "Reduced adenosine-to-inosine miR-455-5p editing promotes melanoma growth and metastasis". Nature Cell Biology 17 (3): 311–321. March 2015. doi:10.1038/ncb3110. PMID 25686251.
- ↑ "Dystonia, mental deterioration, and dyschromatosis symmetrica hereditaria in a family with ADAR1 mutation". Movement Disorders 21 (9): 1510–1513. September 2006. doi:10.1002/mds.21011. PMID 16817193.
- ↑ "Adenosine deaminase acting on RNA-1 (ADAR1) inhibits HIV-1 replication in human alveolar macrophages". PLOS ONE 9 (10). 2014. doi:10.1371/journal.pone.0108476. PMID 25272020. Bibcode: 2014PLoSO...9j8476W.
- ↑ 37.0 37.1 "Enhancement of replication of RNA viruses by ADAR1 via RNA editing and inhibition of RNA-activated protein kinase". Journal of Virology 85 (17): 8460–8466. September 2011. doi:10.1128/JVI.00240-11. PMID 21490091.
- ↑ 38.0 38.1 38.2 "Adenosine Deaminases Acting on RNA (ADARs) and Viral Infections". Annual Review of Virology 8 (1): 239–264. September 2021. doi:10.1146/annurev-virology-091919-065320. PMID 33882257.
- ↑ "Clonal expansion of hypermutated measles virus in a SSPE brain". Virology 197 (1): 188–195. November 1993. doi:10.1006/viro.1993.1579. PMID 8212553.
- ↑ "Biased hypermutation and other genetic changes in defective measles viruses in human brain infections". Cell 55 (2): 255–265. October 1988. doi:10.1016/0092-8674(88)90048-7. PMID 3167982.
- ↑ "Multiple functions of the IKK-related kinase IKKepsilon in interferon-mediated antiviral immunity". Science 315 (5816): 1274–1278. March 2007. doi:10.1126/science.1136567. PMID 17332413.
- ↑ "A-to-G hypermutation in the genome of lymphocytic choriomeningitis virus". Journal of Virology 81 (2): 457–464. January 2007. doi:10.1128/jvi.00067-06. PMID 17020943.
- ↑ "Nuclear antisense RNA induces extensive adenosine modifications and nuclear retention of target transcripts". Proceedings of the National Academy of Sciences of the United States of America 94 (8): 3542–3547. April 1997. doi:10.1073/pnas.94.8.3542. PMID 9108012. Bibcode: 1997PNAS...94.3542K.
- ↑ "A specific base transition occurs on replicating hepatitis delta virus RNA". Journal of Virology 64 (3): 1021–1027. March 1990. doi:10.1128/JVI.64.3.1021-1027.1990. PMID 2304136.
- ↑ "New antiviral pathway that mediates hepatitis C virus replicon interferon sensitivity through ADAR1". Journal of Virology 79 (10): 6291–6298. May 2005. doi:10.1128/JVI.79.10.6291-6298.2005. PMID 15858013.
- ↑ "RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR". The Journal of Biological Chemistry 284 (43): 29350–29356. October 2009. doi:10.1074/jbc.M109.045146. PMID 19710021.
Further reading
- "HIV-1 Envelope gp120 and Viral Particles Block Adenosine Deaminase Binding to Human CD26". Cellular Peptidases in Immune Functions and Diseases. Advances in Experimental Medicine and Biology. 421. 1997. pp. 185–92. doi:10.1007/978-1-4757-9613-1_24. ISBN 978-1-4757-9615-5.
- "Cloning and chromosomal location of human genes inducible by type I interferon". Somatic Cell and Molecular Genetics 14 (5): 415–426. September 1988. doi:10.1007/BF01534709. PMID 3175763.
- "Genomic organization and chromosomal location of the human dsRNA adenosine deaminase gene: the enzyme for glutamate-activated ion channel RNA editing". Journal of Molecular Biology 254 (2): 184–195. November 1995. doi:10.1006/jmbi.1995.0610. PMID 7490742.
- "Expression and regulation by interferon of a double-stranded-RNA-specific adenosine deaminase from human cells: evidence for two forms of the deaminase". Molecular and Cellular Biology 15 (10): 5376–5388. October 1995. doi:10.1128/mcb.15.10.5376. PMID 7565688.
- "Mechanism of interferon action: double-stranded RNA-specific adenosine deaminase from human cells is inducible by alpha and gamma interferons". Virology 210 (2): 508–511. July 1995. doi:10.1006/viro.1995.1370. PMID 7618288.
- "Cloning of cDNAs encoding mammalian double-stranded RNA-specific adenosine deaminase". Molecular and Cellular Biology 15 (3): 1389–1397. March 1995. doi:10.1128/mcb.15.3.1389. PMID 7862132.
- "The interferon-inducible, double-stranded RNA-specific adenosine deaminase gene (DSRAD) maps to human chromosome 1q21.1-21.2". Genomics 30 (2): 372–375. November 1995. doi:10.1006/geno.1995.0034. PMID 8586444.
- "Functionally distinct double-stranded RNA-binding domains associated with alternative splice site variants of the interferon-inducible double-stranded RNA-specific adenosine deaminase". The Journal of Biological Chemistry 272 (7): 4419–4428. February 1997. doi:10.1074/jbc.272.7.4419. PMID 9020165.
- "Adenosine deaminase binding to human CD26 is inhibited by HIV-1 envelope glycoprotein gp120 and viral particles". Journal of Immunology 158 (8): 3721–3729. April 1997. doi:10.4049/jimmunol.158.8.3721. PMID 9103436.
- "A Z-DNA binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase". Proceedings of the National Academy of Sciences of the United States of America 94 (16): 8421–8426. August 1997. doi:10.1073/pnas.94.16.8421. PMID 9237992. Bibcode: 1997PNAS...94.8421H.
- "Double-stranded RNA-specific adenosine deaminase: nucleic acid binding properties". Methods 15 (3): 199–205. July 1998. doi:10.1006/meth.1998.0624. PMID 9735305.
- "Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible". Proceedings of the National Academy of Sciences of the United States of America 96 (8): 4621–4626. April 1999. doi:10.1073/pnas.96.8.4621. PMID 10200312. Bibcode: 1999PNAS...96.4621G.
- "Crystal structure of the Zalpha domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA". Science 284 (5421): 1841–1845. June 1999. doi:10.1126/science.284.5421.1841. PMID 10364558.
- "The solution structure of the Zalpha domain of the human RNA editing enzyme ADAR1 reveals a prepositioned binding surface for Z-DNA". Proceedings of the National Academy of Sciences of the United States of America 96 (22): 12465–12470. October 1999. doi:10.1073/pnas.96.22.12465. PMID 10535945. Bibcode: 1999PNAS...9612465S.
- "The HIV-1 gp120 inhibits the binding of adenosine deaminase to CD26 by a mechanism modulated by CD4 and CXCR4 expression". FEBS Letters 477 (1–2): 123–128. July 2000. doi:10.1016/S0014-5793(00)01751-8. PMID 10899322. Bibcode: 2000FEBSL.477..123B.
- "Comodulation of CXCR4 and CD26 in human lymphocytes". The Journal of Biological Chemistry 276 (22): 19532–19539. June 2001. doi:10.1074/jbc.M004586200. PMID 11278278.
- "Substrate recognition by ADAR1 and ADAR2". RNA 7 (6): 846–858. June 2001. doi:10.1017/S135583820101007X. PMID 11421361.
- "The human but not the Xenopus RNA-editing enzyme ADAR1 has an atypical nuclear localization signal and displays the characteristics of a shuttling protein". Molecular Biology of the Cell 12 (7): 1911–1924. July 2001. doi:10.1091/mbc.12.7.1911. PMID 11451992.
- "Adenosine deaminase acting on RNA 1 limits RIG-I RNA detection and suppresses IFN production responding to viral and endogenous RNAs". Journal of Immunology 193 (7): 3436–3445. October 2014. doi:10.4049/jimmunol.1401136. PMID 25172485.
External links
- OMIM entries on Dyschromatosis Symmetrica Hereditaria 1
- ADAR human gene location in the UCSC Genome Browser.
- ADAR human gene details in the UCSC Genome Browser.
PDB gallery | |
|---|---|
|





 |  |
 EncycloReader
is supported by the
EncycloReader
is supported by the