Table of Contents Categories
Indole
Topic: Chemistry
 From HandWiki - Reading time: 20 min
From HandWiki - Reading time: 20 min
Indole is an organic compound with the formula C
6H
4CCNH
3. Indole is classified as an aromatic heterocycle. It has a bicyclic structure, consisting of a six-membered benzene ring fused to a five-membered pyrrole ring. Indoles are derivatives of indole where one or more of the hydrogen atoms have been replaced by substituent groups. Indoles are widely distributed in nature, most notably as amino acid tryptophan and neurotransmitter serotonin.[1]. In plants, IAA is a common plant hormone which is a derivative of Indole.
General properties and occurrence
Indole is a solid at room temperature. It occurs naturally in human feces and has an intense fecal odor.[2] At very low concentrations, however, it has a flowery smell,[3][4] and is a constituent of many perfumes. It also occurs in coal tar. It has been identified in cannabis.[5] It is the main volatile compound in stinky tofu.[6]
When indole is a substituent on a larger molecule, it is called an indolyl group by systematic nomenclature.
Indole undergoes electrophilic substitution, mainly at position 3 (see diagram in right margin). Substituted indoles are structural elements of (and for some compounds, the synthetic precursors for) the tryptophan-derived tryptamine alkaloids, which includes the neurotransmitter serotonin and the hormone[7] melatonin, as well as the naturally occurring psychedelic drugs dimethyltryptamine and psilocybin. Other indolic compounds include the plant hormone auxin (indole-3-acetic acid, IAA), tryptophol, the anti-inflammatory drug indomethacin, and the betablocker pindolol.
The name indole is a portmanteau of the words indigo and oleum, since indole was first isolated by treatment of the indigo dye with oleum.
History

Indole chemistry began to develop with the study of the dye indigo. Indigo can be converted to isatin and then to oxindole. In 1866, Adolf von Baeyer reduced oxindole to indole using zinc dust.[8] In 1869, he proposed a formula for indole.[9]
Certain indole derivatives were important dyestuffs until the end of the 19th century. In the 1930s, interest in indole intensified when it became known that the indole substituent is present in many important alkaloids, known as indole alkaloids (e.g., tryptophan and auxins), and it remains an active area of research today.[10]
Biosynthesis and function
Indole is biosynthesized in the shikimate pathway via anthranilate.[1] It is an intermediate in the biosynthesis of tryptophan, where it stays inside the tryptophan synthase molecule between the removal of 3-phospho-glyceraldehyde and the condensation with serine. When indole is needed in the cell, it is usually produced from tryptophan by tryptophanase.[11]
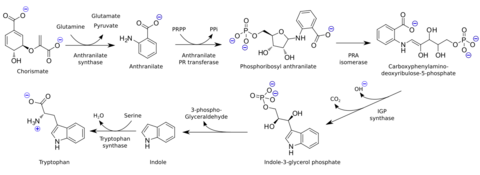
Indole is produced via anthranilate and reacts further to give the amino acid tryptophan.
.svg)
As an intercellular signal molecule, indole regulates various aspects of bacterial physiology, including spore formation, plasmid stability, resistance to drugs, biofilm formation, and virulence.[12] A number of indole derivatives have important cellular functions, including neurotransmitters such as serotonin.[1] {{Annotated image 4 | image = Microbiota-derived 3-Indolepropionic acid-notext.svg | link = Commons:File:Microbiota-derived 3-Indolepropionic acid.svg | header = Tryptophan metabolism by human gastrointestinal microbiota ( ) | header_align = center | header_background = #F0F8FF | align = left | image-width = 600 | image-left = 0 | image-top = 10 | width = 580 | height = 470 | alt = Tryptophan metabolism diagram | caption = {{{caption|This diagram shows the biosynthesis of bioactive compounds (indole and certain other derivatives) from by bacteria in the gut.[13] Indole is produced from tryptophan by bacteria that express tryptophanase.[13] Clostridium sporogenes metabolizes tryptophan into indole and subsequently 3-indolepropionic acid (IPA),[14] a highly potent neuroprotective antioxidant that scavenges hydroxyl radicals.[13][15][16] IPA binds to the pregnane X receptor (PXR) in intestinal cells, thereby facilitating mucosal homeostasis and barrier function.[13] Following absorption from the intestine and distribution to the brain, IPA confers a neuroprotective effect against cerebral ischemia and Alzheimer's disease.[13] Lactobacillus species metabolize tryptophan into {{when pagename is|Indole-3-carboxaldehyde=indole-3-carboxaldehyde|other=indole-3-aldehyde}} (I3A) which acts on the aryl hydrocarbon receptor (AhR) in intestinal immune cells, in turn increasing interleukin-22 (IL-22) production.[13] Indole itself triggers the secretion of glucagon-like peptide-1 (GLP-1) in intestinal L cells and acts as a ligand for AhR.[13] Indole can also be metabolized by the liver into ]], a compound that is toxic in high concentrations and associated with vascular disease and renal dysfunction.[13] AST-120 (activated charcoal), an intestinal sorbent that is [[Oral administrat[[Physics:taken by mouth,Chemistry:Adsorption|adsorbs i]]ndole, in turn decreasing the concentration of indoxyl sulfate in blood plasma.[13] }}} | annot-font-size = 14 | annot-text-align = left | annotations =
expressing
bacteria
immune
cells
↓Activation of glial cells and astrocytes
↓4-Hydroxy-2-nonenal levels
↓DNA damage
–Antioxidant
–Inhibits β-amyloid fibril formation
↑IL-22 production
↑Oxidative stress
↑Smooth muscle cell proliferation
↑Aortic wall thickness and calcification
↑Renal dysfunction
–Uremic toxin
}}
Detection methods
Common classical methods applied for the detection of extracellular and environmental indoles, are Salkowski, Kovács, Ehrlich's reagent assays and HPLC.[17][18][19] For intracellular indole detection and measurement genetically encoded indole-responsive biosensor is applicable.[20]
Medical applications
Indoles and their derivatives are promising against tuberculosis, malaria, diabetes, cancer, migraines, convulsions, hypertension, bacterial infections of methicillin-resistant Staphylococcus aureus (MRSA) and even viruses.[21][22][23][24][25]
Synthetic routes
Indole and its derivatives can also be synthesized by a variety of methods.[26][27][28] According to a 2011 review, all known syntheses fall into 9 categories.[29]
The main industrial routes start from aniline via vapor-phase reaction with ethylene glycol in the presence of catalysts:

In general, reactions are conducted between 200 and 500 °C. Yields can be as high as 60%. Other precursors to indole include formyltoluidine, 2-ethylaniline, and 2-(2-nitrophenyl)ethanol, all of which undergo cyclizations.[30]
Leimgruber–Batcho indole synthesis

The Leimgruber–Batcho indole synthesis is an efficient method of synthesizing indole and substituted indoles.[31] Originally disclosed in a patent in 1976, this method is high-yielding and can generate substituted indoles. This method is especially popular in the pharmaceutical industry, where many pharmaceutical drugs are made up of specifically substituted indoles.
Fischer indole synthesis


One of the oldest and most reliable methods for synthesizing substituted indoles is the Fischer indole synthesis, developed in 1883 by Emil Fischer. Although the synthesis of indole itself is problematic using the Fischer indole synthesis, it is often used to generate indoles substituted in the 2- and/or 3-positions. Indole can still be synthesized, however, using the Fischer indole synthesis by reacting phenylhydrazine with pyruvic acid followed by decarboxylation of the formed indole-2-carboxylic acid. This has also been accomplished in a one-pot synthesis using microwave irradiation.[32]
Other indole-forming reactions
- Bartoli indole synthesis
- Bischler–Möhlau indole synthesis
- Cadogan-Sundberg indole synthesis
- Fukuyama indole synthesis
- Gassman indole synthesis
- Hemetsberger indole synthesis
- Larock indole synthesis
- Madelung synthesis
- Nenitzescu indole synthesis
- Reissert indole synthesis
- Baeyer–Emmerling indole synthesis
- In the Diels–Reese reaction[33][34] dimethyl acetylenedicarboxylate reacts with 1,2-diphenylhydrazine to an adduct, which in xylene gives dimethyl indole-2,3-dicarboxylate and aniline. With other solvents, other products are formed: with glacial acetic acid a pyrazolone, and with pyridine a quinoline.
Chemical reactions of indole
Basicity
Unlike most amines, indole is not basic: just like pyrrole, the aromatic character of the ring means that the lone pair of electrons on the nitrogen atom is not available for protonation.[35] Strong acids such as hydrochloric acid can, however, protonate indole. Indole is primarily protonated at the C3, rather than N1, owing to the enamine-like reactivity of the portion of the molecule located outside of the benzene ring. The protonated form has a pKa of −3.6. The sensitivity of many indolic compounds (e.g., tryptamines) under acidic conditions is caused by this protonation.
Electrophilic substitution
The most reactive position on indole for electrophilic aromatic substitution is C3, which is 1013 times more reactive than benzene. For example, it is alkylated by phosphorylated serine in the biosynthesis of the amino acid tryptophan. Vilsmeier–Haack formylation of indole[36] will take place at room temperature exclusively at C3.

Since the pyrrolic ring is the most reactive portion of indole, electrophilic substitution of the carbocyclic (benzene) ring generally takes place only after N1, C2, and C3 are substituted. A noteworthy exception occurs when electrophilic substitution is carried out in conditions sufficiently acidic to exhaustively protonate C3. In this case, C5 is the most common site of electrophilic attack.[37]
Gramine, a useful synthetic intermediate, is produced via a Mannich reaction of indole with dimethylamine and formaldehyde. It is the precursor to indole-3-acetic acid and synthetic tryptophan.

N–H acidity and organometallic indole anion complexes
The N–H center has a pKa of 21 in DMSO, so that very strong bases such as sodium hydride or n-butyl lithium and water-free conditions are required for complete deprotonation. The resulting organometalic derivatives can react in two ways. The more ionic salts such as the sodium or potassium compounds tend to react with electrophiles at nitrogen-1, whereas the more covalent magnesium compounds (indole Grignard reagents) and (especially) zinc complexes tend to react at carbon 3 (see figure below). In analogous fashion, polar aprotic solvents such as DMF and DMSO tend to favour attack at the nitrogen, whereas nonpolar solvents such as toluene favour C3 attack.[38][39]

Carbon acidity and C2 lithiation
After the N–H proton, the hydrogen at C2 is the next most acidic proton on indole. Reaction of N-protected indoles with butyl lithium or lithium diisopropylamide results in lithiation exclusively at the C2 position. This strong nucleophile can then be used as such with other electrophiles.

Bergman and Venemalm developed a technique for lithiating the 2-position of unsubstituted indole,[40] as did Katritzky.[41]
Oxidation of indole
Due to the electron-rich nature of indole, it is easily oxidized. Simple oxidants such as N-bromosuccinimide will selectively oxidize indole 1 to oxindole (4 and 5).

Cycloadditions of indole
Only the C2–C3 pi bond of indole is capable of cycloaddition reactions. Intramolecular variants are often higher-yielding than intermolecular cycloadditions. For example, Padwa et al.[42] have developed this Diels-Alder reaction to form advanced strychnine intermediates. In this case, the 2-aminofuran is the diene, whereas the indole is the dienophile. Indoles also undergo intramolecular [2+3] and [2+2] cycloadditions.

Despite mediocre yields, intermolecular cycloadditions of indole derivatives have been well documented.[43][44][45][46] One example is the Pictet-Spengler reaction between tryptophan derivatives and aldehydes,[47] which produces a mixture of diastereomers, leading to reduced yield of the desired product.
Hydrogenation
Indoles are susceptible to hydrogenation of the imine subunit[48] to give indolines.

See also
- Indole-3-butyric acid
- Indole test
- Isoindole
- Isoindoline
- Skatole (3-methylindole)
References
- ↑ 1.0 1.1 1.2 Nelson, David L.; Cox, Michael M. (2005), Principles of Biochemistry (4th ed.), New York: W. H. Freeman, ISBN 0-7167-4339-6
- ↑ "indole". Scents and Flavors. https://scentsandflavors.com/database/9dbb4fa2-1957-49cc-ae68-f28f8d93f0a4.
- ↑ "indole". Scents and Flavors. https://scentsandflavors.com/database/9dbb4fa2-1957-49cc-ae68-f28f8d93f0a4.
- ↑ Purves, Dale; Augustine, George J; Fitzpatrick, David; Katz, Lawrence C; LaMantia, Anthony-Samuel; McNamara, James O; Williams, S Mark. "Olfactory Perception in Humans". https://www.ncbi.nlm.nih.gov/books/NBK11032/.
- ↑ Oswald, Iain W. H.; Paryani, Twinkle R.; Sosa, Manuel E.; Ojeda, Marcos A.; Altenbernd, Mark R.; Grandy, Jonathan J.; Shafer, Nathan S.; Ngo, Kim et al. (2023-10-12). "Minor, Nonterpenoid Volatile Compounds Drive the Aroma Differences of Exotic Cannabis" (in en). ACS Omega 8 (42): 39203–39216. doi:10.1021/acsomega.3c04496. ISSN 2470-1343. PMID 37901519.
- ↑ Liu, Yuping; Miao, Zhiwei; Guan, Wei; Sun, Baoguo (26 March 2012). "Analysis of Organic Volatile Flavor Compounds in Fermented Stinky Tofu Using SPME with Different Fiber Coatings". Molecules 17 (4): 3708–3722. doi:10.3390/molecules17043708. PMID 22450681.
- ↑ Lee, Jung Goo (21 October 2019). "The Neuroprotective Effects of Melatonin: Possible Role in the Pathophysiology of Neuropsychiatric Disease". Brain Sciences 9 (285): 285. doi:10.3390/brainsci9100285. PMID 31640239.
- ↑ Baeyer, A. (1866). "Ueber die Reduction aromatischer Verbindungen mittelst Zinkstaub". Annalen der Chemie und Pharmacie 140 (3): 295–296. doi:10.1002/jlac.18661400306. https://archive.org/stream/annalenderchemi11liebgoog#page/n690/mode/2up.
- ↑ Baeyer, A.; Emmerling, A. (1869). "Synthese des Indols". Berichte der Deutschen Chemischen Gesellschaft 2: 679–682. doi:10.1002/cber.186900201268. http://babel.hathitrust.org/cgi/pt?id=uc1.b3481747;view=1up;seq=709.
- ↑ Van Order, R. B.; Lindwall, H. G. (1942). "Indole". Chem. Rev. 30: 69–96. doi:10.1021/cr60095a004.
- ↑ Stephanopoulos, George; Aristidou, Aristos A.; Nielsen, Jens (1998-10-17) (in en). Metabolic Engineering: Principles and Methodologies. Academic Press. p. 251. ISBN 978-0-08-053628-6. https://books.google.com/books?id=9mGzkso4NVQC&pg=PA251.
- ↑ Lee, Jin-Hyung; Lee, Jintae (2010). "Indole as an intercellular signal in microbial communities". FEMS Microbiology Reviews 34 (4): 426–44. doi:10.1111/j.1574-6976.2009.00204.x. ISSN 0168-6445. PMID 20070374.
- ↑ 13.0 13.1 13.2 13.3 13.4 13.5 13.6 13.7 13.8 "Microbial metabolism of dietary components to bioactive metabolites: opportunities for new therapeutic interventions". Genome Med 8 (1): 46. April 2016. doi:10.1186/s13073-016-0296-x. PMID 27102537. "Lactobacillus spp. convert tryptophan to indole-3-aldehyde (I3A) through unidentified enzymes [125]. Clostridium sporogenes convert tryptophan to IPA [6], likely via a tryptophan deaminase. ... IPA also potently scavenges hydroxyl radicals".
Table 2: Microbial metabolites: their synthesis, mechanisms of action, and effects on health and disease
Figure 1: Molecular mechanisms of action of indole and its metabolites on host physiology and disease - ↑ "Metabolomics analysis reveals large effects of gut microflora on mammalian blood metabolites". Proc. Natl. Acad. Sci. U.S.A. 106 (10): 3698–3703. March 2009. doi:10.1073/pnas.0812874106. PMID 19234110. "Production of IPA was shown to be completely dependent on the presence of gut microflora and could be established by colonization with the bacterium Clostridium sporogenes.".
IPA metabolism diagram - ↑ "3-Indolepropionic acid". University of Alberta. http://www.hmdb.ca/metabolites/HMDB02302. Retrieved 12 June 2018. "Indole-3-propionate (IPA), a deamination product of tryptophan formed by symbiotic bacteria in the gastrointestinal tract of mammals and birds. 3-Indolepropionic acid has been shown to prevent oxidative stress and death of primary neurons and neuroblastoma cells exposed to the amyloid beta-protein in the form of amyloid fibrils, one of the most prominent neuropathologic features of Alzheimer's disease. 3-Indolepropionic acid also shows a strong level of neuroprotection in two other paradigms of oxidative stress. (PMID 10419516) ... More recently it has been found that higher indole-3-propionic acid levels in serum/plasma are associated with reduced likelihood of type 2 diabetes and with higher levels of consumption of fiber-rich foods (PMID 28397877)
Origin: • Endogenous • Microbial" - ↑ "Potent neuroprotective properties against the Alzheimer beta-amyloid by an endogenous melatonin-related indole structure, indole-3-propionic acid". J. Biol. Chem. 274 (31): 21937–21942. July 1999. doi:10.1074/jbc.274.31.21937. PMID 10419516. "[Indole-3-propionic acid (IPA)] has previously been identified in the plasma and cerebrospinal fluid of humans, but its functions are not known. ... In kinetic competition experiments using free radical-trapping agents, the capacity of IPA to scavenge hydroxyl radicals exceeded that of melatonin, an indoleamine considered to be the most potent naturally occurring scavenger of free radicals. In contrast with other antioxidants, IPA was not converted to reactive intermediates with pro-oxidant activity.".
- ↑ Ehmann, Axel (1977-02-11). "The van URK-Salkowski reagent — a sensitive and specific chromogenic reagent for silica gel thin-layer chromatographic detection and identification of indole derivatives" (in en). Journal of Chromatography A 132 (2): 267–276. doi:10.1016/S0021-9673(00)89300-0. ISSN 0021-9673. PMID 188858. https://www.sciencedirect.com/science/article/pii/S0021967300893000.
- ↑ Darkoh, Charles; Chappell, Cynthia; Gonzales, Christopher; Okhuysen, Pablo (December 2015). Schloss, P. D.. ed. "A Rapid and Specific Method for the Detection of Indole in Complex Biological Samples" (in en). Applied and Environmental Microbiology 81 (23): 8093–8097. doi:10.1128/AEM.02787-15. ISSN 0099-2240. PMID 26386049. Bibcode: 2015ApEnM..81.8093D.
- ↑ Gilbert, Sarah; Xu, Jenny; Acosta, Kenneth; Poulev, Alexander; Lebeis, Sarah; Lam, Eric (2018). "Bacterial Production of Indole Related Compounds Reveals Their Role in Association Between Duckweeds and Endophytes". Frontiers in Chemistry 6: 265. doi:10.3389/fchem.2018.00265. ISSN 2296-2646. PMID 30050896. Bibcode: 2018FrCh....6..265G.
- ↑ Matulis, Paulius; Kutraite, Ingrida; Augustiniene, Ernesta; Valanciene, Egle; Jonuskiene, Ilona; Malys, Naglis (January 2022). "Development and Characterization of Indole-Responsive Whole-Cell Biosensor Based on the Inducible Gene Expression System from Pseudomonas putida KT2440" (in en). International Journal of Molecular Sciences 23 (9): 4649. doi:10.3390/ijms23094649. ISSN 1422-0067. PMID 35563040.
- ↑ Ramesh, Deepthi; Joji, Annu; Vijayakumar, Balaji Gowrivel; Sethumadhavan, Aiswarya; Mani, Maheswaran; Kannan, Tharanikkarasu (15 July 2020). "Indole chalcones: Design, synthesis, in vitro and in silico evaluation against Mycobacterium tuberculosis" (in en). European Journal of Medicinal Chemistry 198. doi:10.1016/j.ejmech.2020.112358. ISSN 0223-5234. PMID 32361610.
- ↑ Qin, Hua-Li; Liu, Jing; Fang, Wan-Yin; Ravindar, L.; Rakesh, K. P. (15 May 2020). "Indole-based derivatives as potential antibacterial activity against methicillin-resistance Staphylococcus aureus (MRSA)" (in en). European Journal of Medicinal Chemistry 194. doi:10.1016/j.ejmech.2020.112245. ISSN 0223-5234. PMID 32220687.
- ↑ Thanikachalam, Punniyakoti Veeraveedu; Maurya, Rahul Kumar; Garg, Vishali; Monga, Vikramdeep (15 October 2019). "An insight into the medicinal perspective of synthetic analogs of indole: A review" (in en). European Journal of Medicinal Chemistry 180: 562–612. doi:10.1016/j.ejmech.2019.07.019. ISSN 0223-5234. PMID 31344615.
- ↑ Kumari, Archana; Singh, Rajesh K. (1 August 2019). "Medicinal chemistry of indole derivatives: Current to future therapeutic prospectives" (in en). Bioorganic Chemistry 89. doi:10.1016/j.bioorg.2019.103021. ISSN 0045-2068. PMID 31176854.
- ↑ Jia, Yanshu; Wen, Xiaoyue; Gong, Yufeng; Wang, Xuefeng (15 August 2020). "Current scenario of indole derivatives with potential anti-drug-resistant cancer activity" (in en). European Journal of Medicinal Chemistry 200. doi:10.1016/j.ejmech.2020.112359. ISSN 0223-5234. PMID 32531682.
- ↑ Gribble, G. W. (2000). "Recent developments in indole ring synthesis—methodology and applications". J. Chem. Soc. Perkin Trans. 1 (7): 1045. doi:10.1039/a909834h.
- ↑ Cacchi, S.; Fabrizi, G. (2005). "Synthesis and Functionalization of Indoles Through Palladium-catalyzed Reactions". Chem. Rev. 105 (7): 2873–2920. doi:10.1021/cr040639b. PMID 16011327.
- ↑ Humphrey, G. R.; Kuethe, J. T. (2006). "Practical Methodologies for the Synthesis of Indoles". Chem. Rev. 106 (7): 2875–2911. doi:10.1021/cr0505270. PMID 16836303.
- ↑ Taber, Douglass F.; Tirunahari, Pavan K. (September 2011). "Indole synthesis: a review and proposed classification" (in en). Tetrahedron 67 (38): 7195–7210. doi:10.1016/j.tet.2011.06.040. PMID 25484459. PMC 4255418. https://linkinghub.elsevier.com/retrieve/pii/S0040402011009112.
- ↑ Collin, Gerd; Höke, Hartmut. "Ullmann's Encyclopedia of Industrial Chemistry". Ullmann's Encyclopedia of Industrial Chemistry. Weinheim: Wiley-VCH. doi:10.1002/14356007.a14_167.
- ↑ "Indol NSP". https://nsp-sun.com/wp-content/uploads/2020/02/indol.pdf.
- ↑ Bratulescu, George (2008). "A new and efficient one-pot synthesis of indoles". Tetrahedron Letters 49 (6): 984. doi:10.1016/j.tetlet.2007.12.015.
- ↑ Diels, Otto; Reese, Johannes (1934). "Synthesen in der hydroaromatischen Reihe. XX. Über die Anlagerung von Acetylen-dicarbonsäureester an Hydrazobenzol". Justus Liebig's Annalen der Chemie 511: 168. doi:10.1002/jlac.19345110114.
- ↑ Huntress, Ernest H.; Bornstein, Joseph; Hearon, William M. (1956). "An Extension of the Diels-Reese Reaction". J. Am. Chem. Soc. 78 (10): 2225. doi:10.1021/ja01591a055. Bibcode: 1956JAChS..78.2225H.
- ↑ Dewick, Paul M. (2013-03-20) (in en). Essentials of Organic Chemistry: For Students of Pharmacy, Medicinal Chemistry and Biological Chemistry. John Wiley & Sons. p. 143. ISBN 978-1-118-68196-1. https://books.google.com/books?id=RrKgfuRwsqsC&pg=PA143.
- ↑ James, P. N.; Snyder, H. R. (1959). "Indole-3-aldehyde". Organic Syntheses 39: 30. doi:10.15227/orgsyn.039.0030. http://www.orgsyn.org/orgsyn/prep.asp?prep=cv4p0539.
- ↑ Noland, W. E.; Rush, K. R.; Smith, L. R. (1966). "Nitration of Indoles. IV. The Nitration of 2-Phenylindole.". J. Org. Chem. 31: 65–69. doi:10.1021/jo01339a013.
- ↑ Ahmad, Shabee (2025). "Indole Derivatives as Anticancer Agent: Recent Developments". JioaVatar. https://jioavatar.com/a1.
- ↑ Heaney, H.; Ley, S. V. (1974). "1-Benzylindole". Organic Syntheses 54: 58. doi:10.15227/orgsyn.054.0058. http://www.orgsyn.org/orgsyn/prep.asp?prep=cv6p0104.
- ↑ Bergman, J.; Venemalm, L. (1992). "Efficient synthesis of 2-chloro-, 2-bromo-, and 2-iodoindole". J. Org. Chem. 57 (8): 2495. doi:10.1021/jo00034a058.
- ↑ Katritzky, Alan R.; Li, Jianqing; Stevens, Christian V. (1995). "Facile Synthesis of 2-Substituted Indoles and Indolo[3,2-b]carbazoles from 2-(Benzotriazol-1-ylmethyl)indole". J. Org. Chem. 60 (11): 3401–3404. doi:10.1021/jo00116a026.
- ↑ Lynch, S. M.; Bur, S. K.; Padwa, A. (2002). "Intramolecular Amidofuran Cycloadditions across an Indole π-Bond: An Efficient Approach to the Aspidosperma and Strychnos ABCE Core". Org. Lett. 4 (26): 4643–5. doi:10.1021/ol027024q. PMID 12489950.
- ↑ Cox, E. D.; Cook, J. M. (1995). "The Pictet-Spengler condensation: a new direction for an old reaction". Chemical Reviews 95 (6): 1797–1842. doi:10.1021/cr00038a004.
- ↑ Gremmen, C.; Willemse, B.; Wanner, M. J.; Koomen, G.-J. (2000). "Enantiopure Tetrahydro-β-carbolines via Pictet–Spengler Reactions with N-Sulfinyl Tryptamines". Org. Lett. 2 (13): 1955–1958. doi:10.1021/ol006034t. PMID 10891200.
- ↑ Larghi, Enrique L.; Amongero, Marcela; Bracca, Andrea B. J.; Kaufman, Teodoro S. (2005). "The intermolecular Pictet–Spengler condensation with chiral carbonyl derivatives in the stereoselective syntheses of optically-active isoquinoline and indole alkaloids". Arkivoc RL-1554K (12): 98–153. doi:10.3998/ark.5550190.0006.c09.
- ↑ Kaufman, Teodoro S. (2005). "Synthesis of Optically-Active Isoquinoline and Indole Alkaloids Employing the Pictet–Spengler Condensation with Removable Chiral Auxiliaries Bound to Nitrogen". in Vicario, J. L.. New Methods for the Asymmetric Synthesis of Nitrogen Heterocycles. Thiruvananthapuram: Research SignPost. pp. 99–147. ISBN 978-81-7736-278-7.
- ↑ Bonnet, D.; Ganesan, A. (2002). "Solid-Phase Synthesis of Tetrahydro-β-carbolinehydantoins via the N-Acyliminium Pictet–Spengler Reaction and Cyclative Cleavage". J. Comb. Chem. 4 (6): 546–548. doi:10.1021/cc020026h. PMID 12425597.
- ↑ Zhu, G.; Zhang, X. Tetrahedron: Asymmetry 1998, 9, 2415.
General references
- Houlihan, W. J., ed (1972). Indoles Part One. New York: Wiley Interscience.[ISBN missing]
- Sundberg, R. J. (1996). Indoles. San Diego: Academic Press. ISBN 978-0-12-676945-6.
- Joule, J. A.; Mills, K. (2000). Heterocyclic Chemistry. Oxford, UK: Blackwell Science. ISBN 978-0-632-05453-4.
- Joule, J. (2000). E. J., Thomas. ed. Science of Synthesis. 10. Stuttgart: Thieme. p. 361. ISBN 978-3-13-112241-4.
- Schoenherr, H.; Leighton, J. L. (2012). "Direct and Highly Enantioselective Iso-Pictet-Spengler Reactions with α-Ketoamides: Access to Underexplored Indole Core Structures". Org. Lett. 14 (10): 2610–3. doi:10.1021/ol300922b. PMID 22540677.
External links
- Synthesis of indoles (overview of recent methods)
- Synthesis and properties of indoles at chemsynthesis.com
Template:Simple aromatic rings
 |  |
 EncycloReader
is supported by the
EncycloReader
is supported by the