Table of Contents Categories
Inflammation
Topic: Medicine
 From HandWiki - Reading time: 34 min
From HandWiki - Reading time: 34 min
Inflammation (from Latin: inflammatio) is part of the biological defence response of body tissues. Inflammatory immunovascular responses can be triggered by a broad range of stimuli, including physical trauma, "dead, damaged, malfunctioning or stressed tissues", pathogens, irritants, toxins, overuse, autoimmunity, allergens, and foreign bodies (e.g. silica and asbestos).[1] The five cardinal signs are heat, pain, redness, swelling, and loss of function (Latin calor, dolor, rubor, tumor, and functio laesa).
Inflammation is a generic response, and therefore is considered a mechanism of innate immunity, not adaptive immunity.[2] It involves immune cells, blood vessels, and molecular mediators. The function of inflammation is to eliminate the initial cause of cell injury, clear out damaged cells and tissues, and initiate tissue repair. Too little inflammation could lead to progressive tissue destruction by the harmful stimulus (e.g. bacteria) and compromise the survival of the organism. However, inflammation can also have negative effects.[3] For instance, too much inflammation, in the form of chronic inflammation, is associated with various diseases, such as hay fever, periodontal disease, atherosclerosis, and osteoarthritis.
Inflammation can be classified as acute or chronic. Acute inflammation is the initial response of the body to harmful stimuli, and is achieved by the increased movement of plasma and leukocytes (in particular granulocytes) from the blood into the injured tissues. A series of biochemical events propagates and matures the inflammatory response, involving the local vascular system, the immune system, and various cells in the injured tissue. Prolonged inflammation, known as chronic inflammation, leads to a progressive shift in the type of cells present at the site of inflammation, such as mononuclear cells, and involves simultaneous destruction and healing of the tissue.
Inflammation has also been classified as Type 1 and Type 2 based on the type of cytokines and helper T cells (Th1 and Th2) involved.[4]
Signs and symptoms
Cardinal signs – acute
| English | Latin |
|---|---|
| Redness | Rubor |
| Swelling | Tumor |
| Heat | Calor |
| Pain | Dolor |
| Loss of function | Functio laesa[lower-alpha 2] |
Inflammation is characterized by five cardinal signs,[7][8] (the traditional names of which come from Latin):
- Dolor (pain)
- Calor (heat)
- Rubor (redness)
- Tumor (swelling)
- Functio laesa (loss of function)[9]
The first four (classical signs) were described by Celsus (c. 30 BC–38 AD).[10]
Pain is due to the release of chemicals such as bradykinin and histamine that stimulate nerve endings.[7] Heat and redness are due to increased blood flow at body core temperature to the inflamed site. Swelling is caused by accumulation of fluid. The fifth sign, loss of function, is believed to have been added later by Galen,[11] Thomas Sydenham[12] or Rudolf Virchow.[13][7][8] Examples of loss of function include pain that inhibits mobility, severe swelling that prevents movement, having a worse sense of smell during a cold, or having difficulty breathing when bronchitis is present.[14][15] Loss of function has multiple causes.[7]
Cardinal signs – chronic
Common signs and symptoms that develop during chronic inflammation are:[16]
- Body pain, arthralgia, myalgia
- Chronic fatigue and insomnia
- Depression, anxiety and mood disorders
- Gastrointestinal complications such as constipation, diarrhea, and acid reflux
- Weight gain or loss
- Frequent infections
Causes
- Burns[17]
- Frostbite
- Physical injury, blunt or penetrating[18]
- Foreign bodies, including splinters, dirt and debris
- Trauma[17]
- Ionizing radiation
Biological:
- Infection by pathogens[17]
- Immune reactions due to hypersensitivity
- Stress
Chemical:[17]
Psychological:
- Excitement[19]
Acute and chronic
| Acute | Chronic | |
|---|---|---|
| Causative agent | Bacterial pathogens, injured tissues | Persistent acute inflammation due to non-degradable pathogens, viral infection, persistent foreign bodies, or autoimmune reactions |
| Major cells involved | neutrophils (primarily), basophils (inflammatory response), and eosinophils (response to helminth worms and parasites), mononuclear cells (monocytes, macrophages) | Mononuclear cells (monocytes, macrophages, lymphocytes, plasma cells), fibroblasts |
| Primary mediators | Vasoactive amines, eicosanoids | IFN-γ and other cytokines, growth factors, reactive oxygen species, hydrolytic enzymes |
| Onset | Immediate | Delayed |
| Duration | Few days | Up to many months, or years |
| Outcomes | Resolution, abscess formation, chronic inflammation | Tissue destruction, fibrosis, necrosis |
Acute
Acute inflammation is a short-term process, usually appearing within a few minutes or hours and begins to cease upon the removal of the injurious stimulus.[13] It involves a coordinated and systemic mobilization response of various immune, endocrine and neurological mediators of acute inflammation. In a normal healthy response, it becomes activated, clears the pathogen and begins a repair process and then ceases.[20]
Acute inflammation occurs immediately upon injury, lasting only a few days.[21] Cytokines and chemokines promote the migration of neutrophils and macrophages to the site of inflammation.[21] Pathogens, allergens, toxins, burns, and frostbite are some of the typical causes of acute inflammation.[21] Toll-like receptors (TLRs) recognize microbial pathogens.[21] Acute inflammation can be a defensive mechanism to protect tissues against injury.[21] Inflammation lasting 2–6 weeks is designated subacute inflammation.[21][16]
Acute process
This section needs more medical references for verification or relies too heavily on primary sources. (April 2023) |


The process of acute inflammation is initiated by resident immune cells already present in the involved tissue, mainly resident macrophages, dendritic cells, histiocytes, Kupffer cells and mast cells. These cells possess surface receptors known as pattern recognition receptors (PRRs), which recognize (i.e., bind) two subclasses of molecules: pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). PAMPs are compounds that are associated with various pathogens, but which are distinguishable from host molecules. DAMPs are compounds that are associated with host-related injury and cell damage.
At the onset of an infection, burn, or other injuries, these cells undergo activation (one of the PRRs recognize a PAMP or DAMP) and release inflammatory mediators responsible for the clinical signs of inflammation. Vasodilation and its resulting increased blood flow causes the redness (rubor) and increased heat (calor). Increased permeability of the blood vessels results in an exudation (leakage) of plasma proteins and fluid into the tissue (edema), which manifests itself as swelling (tumor). Some of the released mediators such as bradykinin increase the sensitivity to pain (hyperalgesia, dolor). The mediator molecules also alter the blood vessels to permit the migration of leukocytes, mainly neutrophils and macrophages, to flow out of the blood vessels (extravasation) and into the tissue. The neutrophils migrate along a chemotactic gradient created by the local cells to reach the site of injury.[13] The loss of function (functio laesa) is probably the result of a neurological reflex in response to pain.
In addition to cell-derived mediators, several acellular biochemical cascade systems—consisting of preformed plasma proteins—act in parallel to initiate and propagate the inflammatory response. These include the complement system activated by bacteria and the coagulation and fibrinolysis systems activated by necrosis (e.g., burn, trauma).[13]
Acute inflammation may be regarded as the first line of defense against injury. Acute inflammatory response requires constant stimulation to be sustained. Inflammatory mediators are short-lived and are quickly degraded in the tissue. Hence, acute inflammation begins to cease once the stimulus has been removed.[13]
Chronic
Chronic inflammation is inflammation that lasts for months or years.[16] Macrophages, lymphocytes, and plasma cells predominate in chronic inflammation, in contrast to the neutrophils that predominate in acute inflammation.[16] Diabetes, cardiovascular disease, allergies, and chronic obstructive pulmonary disease are examples of diseases mediated by chronic inflammation.[16] Obesity, smoking, stress and insufficient diet are some of the factors that promote chronic inflammation.[16]
Vascular component
This section needs more medical references for verification or relies too heavily on primary sources. (March 2021) |
Vasodilation and increased permeability
As defined, acute inflammation is an immunovascular response to inflammatory stimuli, which can include infection or trauma.[23][24] This means acute inflammation can be broadly divided into a vascular phase that occurs first, followed by a cellular phase involving immune cells (more specifically myeloid granulocytes in the acute setting).[23] The vascular component of acute inflammation involves the movement of plasma fluid, containing important proteins such as fibrin and immunoglobulins (antibodies), into inflamed tissue.
Upon contact with PAMPs, tissue macrophages and mastocytes release vasoactive amines such as histamine and serotonin, as well as eicosanoids such as prostaglandin E2 and leukotriene B4 to remodel the local vasculature.[25] Macrophages and endothelial cells release nitric oxide.[26] These mediators vasodilate and permeabilize the blood vessels, which results in the net distribution of blood plasma from the vessel into the tissue space. The increased collection of fluid into the tissue causes it to swell (edema).[25] This exuded tissue fluid contains various antimicrobial mediators from the plasma such as complement, lysozyme, antibodies, which can immediately deal damage to microbes, and opsonise the microbes in preparation for the cellular phase. If the inflammatory stimulus is a lacerating wound, exuded platelets, coagulants, plasmin and kinins can clot the wounded area using vitamin K-dependent mechanisms[27] and provide haemostasis in the first instance. These clotting mediators also provide a structural staging framework at the inflammatory tissue site in the form of a fibrin lattice – as would construction scaffolding at a construction site – for the purpose of aiding phagocytic debridement and wound repair later on. Some of the exuded tissue fluid is also funneled by lymphatics to the regional lymph nodes, flushing bacteria along to start the recognition and attack phase of the adaptive immune system.

Acute inflammation is characterized by marked vascular changes, including vasodilation, increased permeability and increased blood flow, which are induced by the actions of various inflammatory mediators.[25] Vasodilation occurs first at the arteriole level, progressing to the capillary level, and brings about a net increase in the amount of blood present, causing the redness and heat of inflammation. Increased permeability of the vessels results in the movement of plasma into the tissues, with resultant stasis due to the increase in the concentration of the cells within blood – a condition characterized by enlarged vessels packed with cells. Stasis allows leukocytes to marginate (move) along the endothelium, a process critical to their recruitment into the tissues. Normal flowing blood prevents this, as the shearing force along the periphery of the vessels moves cells in the blood into the middle of the vessel.
Plasma cascade systems
- The complement system, when activated, creates a cascade of chemical reactions that promotes opsonization, chemotaxis, and agglutination, and produces the MAC.
- The kinin system generates proteins capable of sustaining vasodilation and other physical inflammatory effects.
- The coagulation system or clotting cascade, which forms a protective protein mesh over sites of injury.
- The fibrinolysis system, which acts in opposition to the coagulation system, to counterbalance clotting and generate several other inflammatory mediators.
Plasma-derived mediators
* non-exhaustive list
| Name | Produced by | Description |
|---|---|---|
| Bradykinin | Kinin system | A vasoactive protein that is able to induce vasodilation, increase vascular permeability, cause smooth muscle contraction, and induce pain. |
| C3 | Complement system | Cleaves to produce C3a and C3b. C3a stimulates histamine release by mast cells, thereby producing vasodilation. C3b is able to bind to bacterial cell walls and act as an opsonin, which marks the invader as a target for phagocytosis. |
| C5a | Complement system | Stimulates histamine release by mast cells, thereby producing vasodilation. It is also able to act as a chemoattractant to direct cells via chemotaxis to the site of inflammation. |
| Factor XII (Hageman Factor) | Liver | A protein that circulates inactively, until activated by collagen, platelets, or exposed basement membranes via conformational change. When activated, it in turn is able to activate three plasma systems involved in inflammation: the kinin system, fibrinolysis system, and coagulation system. |
| Membrane attack complex | Complement system | A complex of the complement proteins C5b, C6, C7, C8, and multiple units of C9. The combination and activation of this range of complement proteins forms the membrane attack complex, which is able to insert into bacterial cell walls and causes cell lysis with ensuing bacterial death. |
| Plasmin | Fibrinolysis system | Able to break down fibrin clots, cleave complement protein C3, and activate Factor XII. |
| Thrombin | Coagulation system | Cleaves the soluble plasma protein fibrinogen to produce insoluble fibrin, which aggregates to form a blood clot. Thrombin can also bind to cells via the PAR1 receptor to trigger several other inflammatory responses, such as production of chemokines and nitric oxide. |
Cellular component
The cellular component involves leukocytes, which normally reside in blood and must move into the inflamed tissue via extravasation to aid in inflammation.[23] Some act as phagocytes, ingesting bacteria, viruses, and cellular debris. Others release enzymatic granules that damage pathogenic invaders. Leukocytes also release inflammatory mediators that develop and maintain the inflammatory response. In general, acute inflammation is mediated by granulocytes, whereas chronic inflammation is mediated by mononuclear cells such as monocytes and lymphocytes.
Leukocyte extravasation


Various leukocytes, particularly neutrophils, are critically involved in the initiation and maintenance of inflammation. These cells must be able to move to the site of injury from their usual location in the blood, therefore mechanisms exist to recruit and direct leukocytes to the appropriate place. The process of leukocyte movement from the blood to the tissues through the blood vessels is known as extravasation and can be broadly divided up into a number of steps:
- Leukocyte margination and endothelial adhesion: The white blood cells within the vessels which are generally centrally located move peripherally towards the walls of the vessels.[28] Activated macrophages in the tissue release cytokines such as IL-1 and TNFα, which in turn leads to production of chemokines that bind to proteoglycans forming gradient in the inflamed tissue and along the endothelial wall.[25] Inflammatory cytokines induce the immediate expression of P-selectin on endothelial cell surfaces and P-selectin binds weakly to carbohydrate ligands on the surface of leukocytes and causes them to "roll" along the endothelial surface as bonds are made and broken. Cytokines released from injured cells induce the expression of E-selectin on endothelial cells, which functions similarly to P-selectin. Cytokines also induce the expression of integrin ligands such as ICAM-1 and VCAM-1 on endothelial cells, which mediate the adhesion and further slow leukocytes down. These weakly bound leukocytes are free to detach if not activated by chemokines produced in injured tissue after signal transduction via respective G protein-coupled receptors that activates integrins on the leukocyte surface for firm adhesion. Such activation increases the affinity of bound integrin receptors for ICAM-1 and VCAM-1 on the endothelial cell surface, firmly binding the leukocytes to the endothelium.
- Migration across the endothelium, known as transmigration, via the process of diapedesis: Chemokine gradients stimulate the adhered leukocytes to move between adjacent endothelial cells. The endothelial cells retract and the leukocytes pass through the basement membrane into the surrounding tissue using adhesion molecules such as ICAM-1.[28]
- Movement of leukocytes within the tissue via chemotaxis: Leukocytes reaching the tissue interstitium bind to extracellular matrix proteins via expressed integrins and CD44 to prevent them from leaving the site. A variety of molecules behave as chemoattractants, for example, C3a or C5a (the anaphylatoxins), and cause the leukocytes to move along a chemotactic gradient towards the source of inflammation.
Phagocytosis
Extravasated neutrophils in the cellular phase come into contact with microbes at the inflamed tissue. Phagocytes express cell-surface endocytic pattern recognition receptors (PRRs) that have affinity and efficacy against non-specific microbe-associated molecular patterns (PAMPs). Most PAMPs that bind to endocytic PRRs and initiate phagocytosis are cell wall components, including complex carbohydrates such as mannans and β-glucans, lipopolysaccharides (LPS), peptidoglycans, and surface proteins. Endocytic PRRs on phagocytes reflect these molecular patterns, with C-type lectin receptors binding to mannans and β-glucans, and scavenger receptors binding to LPS.
Upon endocytic PRR binding, actin-myosin cytoskeletal rearrangement adjacent to the plasma membrane occurs in a way that endocytoses the plasma membrane containing the PRR-PAMP complex, and the microbe. Phosphatidylinositol and Vps34-Vps15-Beclin1 signalling pathways have been implicated to traffic the endocytosed phagosome to intracellular lysosomes, where fusion of the phagosome and the lysosome produces a phagolysosome. The reactive oxygen species, superoxides and hypochlorite bleach within the phagolysosomes then kill microbes inside the phagocyte.
Phagocytic efficacy can be enhanced by opsonization. Plasma derived complement C3b and antibodies that exude into the inflamed tissue during the vascular phase bind to and coat the microbial antigens. As well as endocytic PRRs, phagocytes also express opsonin receptors Fc receptor and complement receptor 1 (CR1), which bind to antibodies and C3b, respectively. The co-stimulation of endocytic PRR and opsonin receptor increases the efficacy of the phagocytic process, enhancing the lysosomal elimination of the infective agent.
Cell-derived mediators
* non-exhaustive list
| Name | Type | Source | Description |
|---|---|---|---|
| Lysosome granules | Enzymes | Granulocytes | These cells contain a large variety of enzymes that perform a number of functions. Granules can be classified as either specific or azurophilic depending upon the contents, and are able to break down a number of substances, some of which may be plasma-derived proteins that allow these enzymes to act as inflammatory mediators. |
| GM-CSF | Glycoprotein | Macrophages, monocytes, T-cells, B-cells, and tissue-resident cells | Elevated GM-CSF has been shown to contribute to inflammation in inflammatory arthritis, osteoarthritis, colitis asthma, obesity, and COVID-19. |
| Histamine | Monoamine | Mast cells and basophils | Stored in preformed granules, histamine is released in response to a number of stimuli. It causes arteriole dilation, increased venous permeability, and a wide variety of organ-specific effects. |
| IFN-γ | Cytokine | T-cells, NK cells | Antiviral, immunoregulatory, and anti-tumour properties. This interferon was originally called macrophage-activating factor, and is especially important in the maintenance of chronic inflammation. |
| IL-6 | Cytokine and Myokine | Macrophages, osteoblasts, adipocytes, and smooth muscle cells (cytokine) Skeletal muscle cells (myokine) | Pro-inflammatory cytokine secreted by macrophages in response to pathogen-associated molecular patterns (PAMPs); pro-inflammatory cytokine secreted by adipocytes, especially in obesity; anti-inflammatory myokine secreted by skeletal muscle cells in response to exercise. |
| IL-8 | Chemokine | Primarily macrophages | Activation and chemoattraction of neutrophils, with a weak effect on monocytes and eosinophils. |
| Leukotriene B4 | Eicosanoid | Leukocytes, cancer cells | Able to mediate leukocyte adhesion and activation, allowing them to bind to the endothelium and migrate across it. In neutrophils, it is also a potent chemoattractant, and is able to induce the formation of reactive oxygen species and the release of lysosomal enzymes by these cells. |
| LTC4, LTD4 | Eicosanoid | eosinophils, mast cells, macrophages | These three Cysteine-containing leukotrienes contract lung airways, increase micro-vascular permeability, stimulate mucus secretion, and promote eosinophil-based inflammation in the lung, skin, nose, eye, and other tissues. |
| 5-oxo-eicosatetraenoic acid | Eicosanoid | Leukocytes, cancer cells | Potent stimulator of neutrophil chemotaxis, lysosome enzyme release, and reactive oxygen species formation; monocyte chemotaxis; and with even greater potency eosinophil chemotaxis, lysosome enzyme release, and reactive oxygen species formation. |
| 5-HETE | Eicosanoid | Leukocytes | Metabolic precursor to 5-Oxo-eicosatetraenoic acid, it is a less potent stimulator of neutrophil chemotaxis, lysosome enzyme release, and reactive oxygen species formation; monocyte chemotaxis; and eosinophil chemotaxis, lysosome enzyme release, and reactive oxygen species formation. |
| Prostaglandins | Eicosanoid | Mast cells | A group of lipids that can cause vasodilation, fever, and pain. |
| Nitric oxide | Soluble gas | Macrophages, endothelial cells, some neurons | Potent vasodilator, relaxes smooth muscle, reduces platelet aggregation, aids in leukocyte recruitment, direct antimicrobial activity in high concentrations. |
| TNF-α and IL-1 | Cytokines | Primarily macrophages | Both affect a wide variety of cells to induce many similar inflammatory reactions: fever, production of cytokines, endothelial gene regulation, chemotaxis, leukocyte adherence, activation of fibroblasts. Responsible for the systemic effects of inflammation, such as loss of appetite and increased heart rate. TNF-α inhibits osteoblast differentiation. |
| Tryptase | Enzymes | Mast Cells | This serine protease is believed to be exclusively stored in mast cells and secreted, along with histamine, during mast cell activation.[29][30][31] |
Morphologic patterns
Specific patterns of acute and chronic inflammation are seen during particular situations that arise in the body, such as when inflammation occurs on an epithelial surface, or pyogenic bacteria are involved.
- Granulomatous inflammation: Characterised by the formation of granulomas, they are the result of a limited but diverse number of diseases, which include among others tuberculosis, leprosy, sarcoidosis, and syphilis.
- Fibrinous inflammation: Inflammation resulting in a large increase in vascular permeability allows fibrin to pass through the blood vessels. If an appropriate procoagulative stimulus is present, such as cancer cells,[13] a fibrinous exudate is deposited. This is commonly seen in serous cavities, where the conversion of fibrinous exudate into a scar can occur between serous membranes, limiting their function. The deposit sometimes forms a pseudomembrane sheet. During inflammation of the intestine (pseudomembranous colitis), pseudomembranous tubes can be formed.
- Purulent inflammation: Inflammation resulting in large amount of pus, which consists of neutrophils, dead cells, and fluid. Infection by pyogenic bacteria such as staphylococci is characteristic of this kind of inflammation. Large, localised collections of pus enclosed by surrounding tissues are called abscesses.
- Serous inflammation: Characterised by the copious effusion of non-viscous serous fluid, commonly produced by mesothelial cells of serous membranes, but may be derived from blood plasma. Skin blisters exemplify this pattern of inflammation.
- Ulcerative inflammation: Inflammation occurring near an epithelium can result in the necrotic loss of tissue from the surface, exposing lower layers. The subsequent excavation in the epithelium is known as an ulcer.
Disorders
.png)

Inflammatory abnormalities are a large group of disorders that underlie a vast variety of human diseases. The immune system is often involved with inflammatory disorders, as demonstrated in both allergic reactions and some myopathies, with many immune system disorders resulting in abnormal inflammation. Non-immune diseases with causal origins in inflammatory processes include cancer, atherosclerosis, and ischemic heart disease.[13]
Examples of disorders associated with inflammation include:
- Acne vulgaris
- Asthma
- Autoimmune diseases
- Autoinflammatory diseases
- Celiac disease
- Chronic prostatitis
- Colitis
- Diverticulitis
- Familial Mediterranean Fever
- Glomerulonephritis
- Hidradenitis suppurativa
- Hypersensitivities
- Inflammatory bowel diseases
- Interstitial cystitis
- Lichen planus
- Mast Cell Activation Syndrome
- Mastocytosis
- Otitis
- Pelvic inflammatory disease
- Peripheral ulcerative keratitis
- Pneumonia
- Reperfusion injury
- Rheumatic fever
- Rheumatoid arthritis
- Rhinitis
- Sarcoidosis
- Transplant rejection
- Vasculitis
Atherosclerosis
Atherosclerosis, formerly considered a lipid storage disorder, is now understood as a chronic inflammatory condition involving the arterial walls.[32] Research has established a fundamental role for inflammation in mediating all stages of atherosclerosis from initiation through progression and, ultimately, the thrombotic complications from it.[32] These new findings reveal links between traditional risk factors like cholesterol levels and the underlying mechanisms of atherogenesis.
Clinical studies have shown that this emerging biology of inflammation in atherosclerosis applies directly to people.[32] For instance, elevation in markers of inflammation predicts outcomes of people with acute coronary syndromes, independently of myocardial damage. In addition, low-grade chronic inflammation, as indicated by levels of the inflammatory marker C-reactive protein, prospectively defines risk of atherosclerotic complications, thus adding to prognostic information provided by traditional risk factors, such as LDL levels.[33][32]
Moreover, certain treatments that reduce coronary risk also limit inflammation. Notably, lipid-lowering medications such as statins have shown anti-inflammatory effects, which may contribute to their efficacy beyond just lowering LDL levels.[34] This emerging understanding of inflammation's role in atherosclerosis has had significant clinical implications, influencing both risk stratification and therapeutic strategies.
Emerging treatments
Recent developments in the treatment of atherosclerosis have focused on addressing inflammation directly. New anti-inflammatory drugs, such as monoclonal antibodies targeting IL-1β, have been studied in large clinical trials, showing promising results in reducing cardiovascular events.[35] These drugs offer a potential new avenue for treatment, particularly for patients who do not respond adequately to statins. However, concerns about long-term safety and cost remain significant barriers to widespread adoption.
Connection to depression
Inflammatory processes can be triggered by negative cognition or their consequences, such as stress, violence, or deprivation. Negative cognition may therefore contribute to inflammation, which in turn can lead to depression. A 2019 meta-analysis found that chronic inflammation is associated with a 30% increased risk of developing major depressive disorder, supporting the link between inflammation and mental health.[36]
Allergy
An allergic reaction, formally known as type 1 hypersensitivity, is the result of an inappropriate immune response triggering inflammation, vasodilation, and nerve irritation. A common example is hay fever, which is caused by a hypersensitive response by mast cells to allergens. Pre-sensitised mast cells respond by degranulating, releasing vasoactive chemicals such as histamine. These chemicals propagate an excessive inflammatory response characterised by blood vessel dilation, production of pro-inflammatory molecules, cytokine release, and recruitment of leukocytes.[13] Severe inflammatory response may mature into a systemic response known as anaphylaxis.
Myopathies
Inflammatory myopathies are caused by the immune system inappropriately attacking components of muscle, leading to signs of muscle inflammation. They may occur in conjunction with other immune disorders, such as systemic sclerosis, and include dermatomyositis, polymyositis, and inclusion body myositis.[13]
Leukocyte defects
Due to the central role of leukocytes in the development and propagation of inflammation, defects in leukocyte functionality often result in a decreased capacity for inflammatory defense with subsequent vulnerability to infection.[13] Dysfunctional leukocytes may be unable to correctly bind to blood vessels due to surface receptor mutations, digest bacteria (Chédiak–Higashi syndrome), or produce microbicides (chronic granulomatous disease). In addition, diseases affecting the bone marrow may result in abnormal or few leukocytes.
Pharmacological
Certain drugs or exogenous chemical compounds are known to affect inflammation. Vitamin A deficiency, for example, causes an increase in inflammatory responses,[37] and anti-inflammatory drugs work specifically by inhibiting the enzymes that produce inflammatory eicosanoids. Additionally, certain illicit drugs such as cocaine and ecstasy may exert some of their detrimental effects by activating transcription factors intimately involved with inflammation (e.g. NF-κB).[38][39]
Cancer
Inflammation orchestrates the microenvironment around tumours, contributing to proliferation, survival and migration.[40] Cancer cells use selectins, chemokines and their receptors for invasion, migration and metastasis.[41] On the other hand, many cells of the immune system contribute to cancer immunology, suppressing cancer.[42] Molecular intersection between receptors of steroid hormones, which have important effects on cellular development, and transcription factors that play key roles in inflammation, such as NF-κB, may mediate some of the most critical effects of inflammatory stimuli on cancer cells.[43] This capacity of a mediator of inflammation to influence the effects of steroid hormones in cells is very likely to affect carcinogenesis. On the other hand, due to the modular nature of many steroid hormone receptors, this interaction may offer ways to interfere with cancer progression, through targeting of a specific protein domain in a specific cell type. Such an approach may limit side effects that are unrelated to the tumor of interest, and may help preserve vital homeostatic functions and developmental processes in the organism.
There is some evidence from 2009 to suggest that cancer-related inflammation (CRI) may lead to accumulation of random genetic alterations in cancer cells.[44][needs update]
Role in cancer
In 1863, Rudolf Virchow hypothesized that the origin of cancer was at sites of chronic inflammation.[41][45] As of 2012, chronic inflammation was estimated to contribute to approximately 15% to 25% of human cancers.[45][46]
Mediators and DNA damage in cancer
An inflammatory mediator is a messenger that acts on blood vessels and/or cells to promote an inflammatory response.[47] Inflammatory mediators that contribute to neoplasia include prostaglandins, inflammatory cytokines such as IL-1β, TNF-α, IL-6 and IL-15 and chemokines such as IL-8 and GRO-alpha.[48][45] These inflammatory mediators, and others, orchestrate an environment that fosters proliferation and survival.[41][48]
Inflammation also causes DNA damages due to the induction of reactive oxygen species (ROS) by various intracellular inflammatory mediators.[41][48][45] In addition, leukocytes and other phagocytic cells attracted to the site of inflammation induce DNA damages in proliferating cells through their generation of ROS and reactive nitrogen species (RNS). ROS and RNS are normally produced by these cells to fight infection.[41] ROS, alone, cause more than 20 types of DNA damage.[49] Oxidative DNA damages cause both mutations[50] and epigenetic alterations.[51][45][52] RNS can also cause mutagenic DNA damages.[53]
A normal cell may undergo carcinogenesis to become a cancer cell if it is frequently subjected to DNA damage during long periods of chronic inflammation. DNA damages may cause genetic mutations due to inaccurate repair. In addition, mistakes in the DNA repair process may cause epigenetic alterations.[45][48][52] Mutations and epigenetic alterations that are replicated and provide a selective advantage during somatic cell proliferation may be carcinogenic.
Genome-wide analyses of human cancer tissues reveal that a single typical cancer cell may possess roughly 100 mutations in coding regions, 10–20 of which are "driver mutations" that contribute to cancer development.[45] However, chronic inflammation also causes epigenetic changes such as DNA methylations, that are often more common than mutations. Typically, several hundreds to thousands of genes are methylated in a cancer cell (see DNA methylation in cancer). Sites of oxidative damage in chromatin can recruit complexes that contain DNA methyltransferases (DNMTs), a histone deacetylase (SIRT1), and a histone methyltransferase (EZH2), and thus induce DNA methylation.[45][54][55] DNA methylation of a CpG island in a promoter region may cause silencing of its downstream gene (see CpG site and regulation of transcription in cancer). DNA repair genes, in particular, are frequently inactivated by methylation in various cancers (see hypermethylation of DNA repair genes in cancer). A 2018 report[56] evaluated the relative importance of mutations and epigenetic alterations in progression to two different types of cancer. This report showed that epigenetic alterations were much more important than mutations in generating gastric cancers (associated with inflammation).[57] However, mutations and epigenetic alterations were of roughly equal importance in generating esophageal squamous cell cancers (associated with tobacco chemicals and acetaldehyde, a product of alcohol metabolism).
HIV and AIDS
It has long been recognized that infection with HIV is characterized not only by development of profound immunodeficiency but also by sustained inflammation and immune activation.[58][59][60] A substantial body of evidence implicates chronic inflammation as a critical driver of immune dysfunction, premature appearance of aging-related diseases, and immune deficiency.[58][61] Many now regard HIV infection not only as an evolving virus-induced immunodeficiency, but also as chronic inflammatory disease.[62] Even after the introduction of effective antiretroviral therapy (ART) and effective suppression of viremia in HIV-infected individuals, chronic inflammation persists. Animal studies also support the relationship between immune activation and progressive cellular immune deficiency: SIVsm infection of its natural nonhuman primate hosts, the sooty mangabey, causes high-level viral replication but limited evidence of disease.[63][64] This lack of pathogenicity is accompanied by a lack of inflammation, immune activation and cellular proliferation. In sharp contrast, experimental SIVsm infection of rhesus macaque produces immune activation and AIDS-like disease with many parallels to human HIV infection.[65]
Delineating how CD4 T cells are depleted and how chronic inflammation and immune activation are induced lies at the heart of understanding HIV pathogenesis—one of the top priorities for HIV research by the Office of AIDS Research, National Institutes of Health. Recent studies demonstrated that caspase-1-mediated pyroptosis, a highly inflammatory form of programmed cell death, drives CD4 T-cell depletion and inflammation by HIV.[66][67][68] These are the two signature events that propel HIV disease progression to AIDS. Pyroptosis appears to create a pathogenic vicious cycle in which dying CD4 T cells and other immune cells (including macrophages and neutrophils) release inflammatory signals that recruit more cells into the infected lymphoid tissues to die. The feed-forward nature of this inflammatory response produces chronic inflammation and tissue injury.[69] Identifying pyroptosis as the predominant mechanism that causes CD4 T-cell depletion and chronic inflammation, provides novel therapeutic opportunities, namely caspase-1 which controls the pyroptotic pathway. In this regard, pyroptosis of CD4 T cells and secretion of pro-inflammatory cytokines such as IL-1β and IL-18 can be blocked in HIV-infected human lymphoid tissues by addition of the caspase-1 inhibitor VX-765,[66] which has already proven to be safe and well tolerated in phase II human clinical trials.[70] These findings could propel development of an entirely new class of "anti-AIDS" therapies that act by targeting the host rather than the virus. Such agents would almost certainly be used in combination with ART. By promoting "tolerance" of the virus instead of suppressing its replication, VX-765 or related drugs may mimic the evolutionary solutions occurring in multiple monkey hosts (e.g. the sooty mangabey) infected with species-specific lentiviruses that have led to a lack of disease, no decline in CD4 T-cell counts, and no chronic inflammation.
Connection to depression
There is evidence for a link between inflammation and depression.[71] Inflammatory processes can be triggered by negative cognitions or their consequences, such as stress, violence, or deprivation. Thus, negative cognitions can cause inflammation that can, in turn, lead to depression.[72][73] In addition, there is increasing evidence that inflammation can cause depression because of the increase of cytokines, setting the brain into a "sickness mode".[74]
Classical symptoms of being physically sick, such as lethargy, show a large overlap in behaviors that characterize depression. Levels of cytokines tend to increase sharply during the depressive episodes of people with bipolar disorder and drop off during remission.[75] Furthermore, it has been shown in clinical trials that anti-inflammatory medicines taken in addition to antidepressants not only significantly improves symptoms but also increases the proportion of subjects positively responding to treatment.[76] Inflammations that lead to serious depression could be caused by common infections such as those caused by a virus, bacteria or even parasites.[77]
Connection to delirium
There is evidence for a link between inflammation and delirium based on the results of a recent longitudinal study investigating CRP in COVID-19 patients.[78]
Systemic effects
An infectious organism can escape the confines of the immediate tissue via the circulatory system or lymphatic system, where it may spread to other parts of the body. If an organism is not contained by the actions of acute inflammation, it may gain access to the lymphatic system via nearby lymph vessels. An infection of the lymph vessels is known as lymphangitis, and infection of a lymph node is known as lymphadenitis. When lymph nodes cannot destroy all pathogens, the infection spreads further. A pathogen can gain access to the bloodstream through lymphatic drainage into the circulatory system.
When inflammation overwhelms the host, systemic inflammatory response syndrome is diagnosed. When it is due to infection, the term sepsis is applied, with the terms bacteremia being applied specifically for bacterial sepsis and viremia specifically to viral sepsis. Vasodilation and organ dysfunction are serious problems associated with widespread infection that may lead to septic shock and death.[79]
Acute-phase proteins
Inflammation also is characterized by high systemic levels of acute-phase proteins. In acute inflammation, these proteins prove beneficial; however, in chronic inflammation, they can contribute to amyloidosis.[13] These proteins include C-reactive protein, serum amyloid A, and serum amyloid P, which cause a range of systemic effects including:[13]
- Fever
- Increased blood pressure
- Decreased sweating
- Malaise
- Loss of appetite
- Somnolence
Leukocyte numbers
Inflammation often affects the numbers of leukocytes present in the body:
- Leukocytosis is often seen during inflammation induced by infection, where it results in a large increase in the amount of leukocytes in the blood, especially immature cells. Leukocyte numbers usually increase to between 15 000 and 20 000 cells per microliter, but extreme cases can see it approach 100 000 cells per microliter.[13] Bacterial infection usually results in an increase of neutrophils, creating neutrophilia, whereas diseases such as asthma, hay fever, and parasite infestation result in an increase in eosinophils, creating eosinophilia.[13]
- Leukopenia can be induced by certain infections and diseases, including viral infection, Rickettsia infection, some protozoa, tuberculosis, and some cancers.[13]
Interleukins and obesity
With the discovery of interleukins (IL), the concept of systemic inflammation developed. Although the processes involved are identical to tissue inflammation, systemic inflammation is not confined to a particular tissue but involves the endothelium and other organ systems.
Chronic inflammation is widely observed in obesity.[80][81] Obese people commonly have many elevated markers of inflammation, including:[82][83]
Low-grade chronic inflammation is characterized by a two- to threefold increase in the systemic concentrations of cytokines such as TNF-α, IL-6, and CRP.[86] Waist circumference correlates significantly with systemic inflammatory response.[87]
Loss of white adipose tissue reduces levels of inflammation markers.[80] As of 2017 the association of systemic inflammation with insulin resistance and type 2 diabetes, and with atherosclerosis was under preliminary research, although rigorous clinical trials had not been conducted to confirm such relationships.[88]
C-reactive protein (CRP) is generated at a higher level in obese people, and may increase the risk for cardiovascular diseases.[89]
Outcomes
The outcome in a particular circumstance will be determined by the tissue in which the injury has occurred—and the injurious agent that is causing it. Here are the possible outcomes to inflammation:[13]
- Resolution
The complete restoration of the inflamed tissue back to a normal status. Inflammatory measures such as vasodilation, chemical production, and leukocyte infiltration cease, and damaged parenchymal cells regenerate. Such is usually the outcome when limited or short-lived inflammation has occurred. - Fibrosis
Large amounts of tissue destruction, or damage in tissues unable to regenerate, cannot be regenerated completely by the body. Fibrous scarring occurs in these areas of damage, forming a scar composed primarily of collagen. The scar will not contain any specialized structures, such as parenchymal cells, hence functional impairment may occur. - Abscess formation
A cavity is formed containing pus, an opaque liquid containing dead white blood cells and bacteria with general debris from destroyed cells. - Chronic inflammation
In acute inflammation, if the injurious agent persists then chronic inflammation will ensue. This process, marked by inflammation lasting many days, months or even years, may lead to the formation of a chronic wound. Chronic inflammation is characterised by the dominating presence of macrophages in the injured tissue. These cells are powerful defensive agents of the body, but the toxins they release—including reactive oxygen species—are injurious to the organism's own tissues as well as invading agents. As a consequence, chronic inflammation is almost always accompanied by tissue destruction.
Resolution
The inflammatory response must be actively terminated when no longer needed to prevent unnecessary "bystander" damage to tissues.[13] Failure to do so results in chronic inflammation, and cellular destruction. Resolution of inflammation occurs by different mechanisms in different tissues. Mechanisms that serve to terminate inflammation include:[13][90]
- Short half-life of inflammatory mediators in vivo.
- Production and release of transforming growth factor (TGF) beta from macrophages[91][92][93]
- Production and release of interleukin 10 (IL-10)[94]
- Production of anti-inflammatory specialized proresolving mediators, i.e. lipoxins, resolvins, maresins, and neuroprotectins[95][96]
- Downregulation of pro-inflammatory molecules, such as leukotrienes.
- Upregulation of anti-inflammatory molecules such as the interleukin 1 receptor antagonist or the soluble tumor necrosis factor receptor (TNFR)
- Apoptosis of pro-inflammatory cells[97]
- Desensitization of receptors.
- Increased survival of cells in regions of inflammation due to their interaction with the extracellular matrix (ECM)[98][99]
- Downregulation of receptor activity by high concentrations of ligands
- Cleavage of chemokines by matrix metalloproteinases (MMPs) might lead to production of anti-inflammatory factors.[100]
| “ | Acute inflammation normally resolves by mechanisms that have remained somewhat elusive. Emerging evidence now suggests that an active, coordinated program of resolution initiates in the first few hours after an inflammatory response begins. After entering tissues, granulocytes promote the switch of arachidonic acid–derived prostaglandins and leukotrienes to lipoxins, which initiate the termination sequence. Neutrophil recruitment thus ceases and programmed death by apoptosis is engaged. These events coincide with the biosynthesis, from omega-3 polyunsaturated fatty acids, of resolvins and protectins, which critically shorten the period of neutrophil infiltration by initiating apoptosis. As a consequence, apoptotic neutrophils undergo phagocytosis by macrophages, leading to neutrophil clearance and release of anti-inflammatory and reparative cytokines such as transforming growth factor-β1. The anti-inflammatory program ends with the departure of macrophages through the lymphatics.[101] | ” |
| — Charles N. Serhan | ||
Examples
Inflammation is usually indicated by adding the suffix "itis", as shown below. However, some conditions, such as asthma and pneumonia, do not follow this convention. More examples are available at List of types of inflammation.
-
 Acute appendicitis
Acute appendicitis -
 Acute dermatitis
Acute dermatitis -
 Acute infective meningitis
Acute infective meningitis -
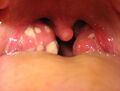 Acute tonsillitis
Acute tonsillitis



See also
Notes
- ↑ All these signs may be observed in specific instances, but no single sign must, as a matter of course, be present.[5] These are the original, or cardinal signs of inflammation.[5]
- ↑ Functio laesa is an antiquated notion, as it is not unique to inflammation and is a characteristic of many disease states.[6]
References
- ↑ Hannoodee, Sally; Nasuruddin, Dian N. (2024-06-08), "Acute Inflammatory Response" (in en), StatPearls [Internet] (StatPearls Publishing), PMID 32310543, https://www.ncbi.nlm.nih.gov/sites/books/NBK556083/, retrieved 2025-10-30
- ↑ "Ch.2 Innate Immunity". Basic Immunology. Functions and disorders of the immune system (3rd ed.). Saunders/Elsevier. 2009. ISBN 978-1-4160-4688-2.
- ↑ "Inflammation and Your Health". https://www.cedars-sinai.org/discoveries/inflammation.html.
- ↑ "Th1 and Th2 responses: what are they?". BMJ 321 (7258): 424. August 2000. doi:10.1136/bmj.321.7258.424. PMID 10938051. PMC 27457. https://www.bmj.com/content/321/7258/424.1. Retrieved 1 July 2021.
- ↑ 5.0 5.1 Stedman's Medical Dictionary (Twenty-fifth ed.). Williams & Wilkins. 1990.
- ↑ "Disturbance of function (functio laesa): the legendary fifth cardinal sign of inflammation, added by Claudius Galen to the four cardinal signs of Celsus". Bulletin of the New York Academy of Medicine 47 (3): 303–22. March 1971. PMID 5276838.
- ↑ 7.0 7.1 7.2 7.3 "Part A. "General Pathology", Section II. "The Host Response to Injury", Chapter 3. "The Acute Inflammatory Response", sub-section "Cardinal Clinical Signs"". Concise Pathology (3rd ed.). McGraw-Hill. 2005. ISBN 978-0-8385-1499-3. OCLC 150148447. http://www.accessmedicine.com/content.aspx?aID=183351. Retrieved 2008-11-05.
- ↑ 8.0 8.1 "Disturbance of function (functio laesa): the legendary fifth cardinal sign of inflammation, added by Galen to the four cardinal signs of Celsus". Bulletin of the New York Academy of Medicine 47 (3): 303–322. March 1971. PMID 5276838.
- ↑ A massage Therapist Guide to Pathology (4th ed.). Wolters Kluwer. 2009. ISBN 978-0-7817-6919-8. http://thepoint.lww.com/Book/ShowWithResource/2931?resourceId=16419. Retrieved 6 October 2010.
- ↑ Brief History of Vision and Ocular Medicine. Kugler Publications. 2009. p. 97. ISBN 978-90-6299-220-1. https://books.google.com/books?id=t_5pzrF1QocC&pg=PA97.
- ↑ Essentials of pahtophysiology: concepts of altered health states. Hagerstown, MD: Lippincott Williams & Wilkins. 2007. pp. 270. ISBN 978-0-7817-7087-3.
- ↑ The worst of evils: man's fight against pain. New Haven, Conn: Yale University Press. 2006. pp. 22. ISBN 978-0-300-11322-8. https://archive.org/details/worstofevilsmans00dorm/page/22.
- ↑ 13.00 13.01 13.02 13.03 13.04 13.05 13.06 13.07 13.08 13.09 13.10 13.11 13.12 13.13 13.14 13.15 13.16 13.17 Robbins Pathologic Basis of Disease. Philadelphia: W.B Saunders Company. 1998. ISBN 978-0-7216-7335-6.
- ↑ InformedHealth.org [Internet]. Institute for Quality and Efficiency in Health Care (IQWiG). 22 February 2018. https://www.ncbi.nlm.nih.gov/books/NBK279298/.
- ↑ "Inflammation | Definition, Symptoms, Treatment, & Facts | Britannica". 11 March 2024. https://www.britannica.com/science/inflammation.
- ↑ 16.0 16.1 16.2 16.3 16.4 16.5 "Chronic inflammation". StatPearls, US National Library of Medicine. 7 August 2023. https://www.ncbi.nlm.nih.gov/books/NBK493173/.
- ↑ 17.0 17.1 17.2 17.3 Guyton and Hall textbook of medical physiology (12th ed.). Philadelphia, Pa.: Saunders/Elsevier. 2011. p. 428. ISBN 978-1-4160-4574-8.
- ↑ "Leukocyte–Endothelial Cell Adhesion". Inflammation and the Microcirculation. Integrated Systems Physiology—From Cell to Function. 2. Morgan & Claypool Life Sciences. 2010. pp. 1–87. doi:10.4199/C00013ED1V01Y201006ISP008. https://www.ncbi.nlm.nih.gov/books/NBK53380/. Retrieved 1 July 2017.
- ↑ "Physiological responses to emotional excitement in healthy subjects and patients with coronary artery disease". Autonomic Neuroscience 177 (2): 280–5. October 2013. doi:10.1016/j.autneu.2013.06.001. PMID 23916871.
- ↑ "The dynamics of acute inflammation". Journal of Theoretical Biology 230 (2): 145–55. September 2004. doi:10.1016/j.jtbi.2004.04.044. PMID 15321710. Bibcode: 2004PhDT.......405K.
- ↑ 21.0 21.1 21.2 21.3 21.4 21.5 "Acute Inflammatory Response". StatPearls. 2020. PMID 32310543. https://www.ncbi.nlm.nih.gov/books/NBK556083/. Retrieved 28 December 2020.
- ↑ Pathologic basis of disease (10th ed.). Philadelphia, PA: Saunders Elsevier. 2020.
- ↑ 23.0 23.1 23.2 "Inflammation and Nutritional Science for Programs/Policies and Interpretation of Research Evidence (INSPIRE)". The Journal of Nutrition 145 (5): 1039S–1108S. May 2015. doi:10.3945/jn.114.194571. PMID 25833893.
- ↑ "Inflammation and immune resolution". Clinical and Experimental Immunology 193 (1): 1–2. July 2018. doi:10.1111/cei.13155. PMID 29987840.
- ↑ 25.0 25.1 25.2 25.3 "Origin and physiological roles of inflammation". Nature 454 (7203): 428–435. July 2008. doi:10.1038/nature07201. PMID 18650913. Bibcode: 2008Natur.454..428M.
- ↑ "Humoral Innate Immunity and Acute-Phase Proteins". The New England Journal of Medicine 388 (5): 439–452. February 2023. doi:10.1056/NEJMra2206346. PMID 36724330.
- ↑ "Vitamin K" (in en). Present Knowledge in Nutrition. Elsevier. 2020. pp. 137–153. doi:10.1016/b978-0-323-66162-1.00008-1. ISBN 978-0-323-66162-1. https://linkinghub.elsevier.com/retrieve/pii/B9780323661621000081. Retrieved 2023-02-17.
- ↑ 28.0 28.1 Muir's Textbook of Pathology (15th ed.). CRC Press. 2014. pp. 59. ISBN 978-1-4441-8499-0.
- ↑ "Role of Interleukin-31 and Oncostatin M in Itch and Neuroimmune Communication". Itch: Mechanisms and Treatment. Frontiers in Neuroscience. Boca Raton (FL): CRC Press/Taylor & Francis. 2014. ISBN 978-1-4665-0543-8. https://www.ncbi.nlm.nih.gov/books/NBK200913/.
- ↑ "Mast cell tryptases and chymases in inflammation and host defense". Immunological Reviews 217 (1): 141–54. June 2007. doi:10.1111/j.1600-065x.2007.00509.x. PMID 17498057.
- ↑ "Mast cell proteases as pharmacological targets". European Journal of Pharmacology. Pharmacological modulation of Mast cells and Basophils 778: 44–55. May 2016. doi:10.1016/j.ejphar.2015.04.045. PMID 25958181.
- ↑ 32.0 32.1 32.2 32.3 "Inflammation during the life cycle of the atherosclerotic plaque". Cardiovascular Research 117 (13): 2525–2536. November 2021. doi:10.1093/cvr/cvab303. PMID 34550337.
- ↑ "Role of inflammation in atherosclerosis". Journal of Nuclear Medicine 48 (11): 1800–1815. November 2007. doi:10.2967/jnumed.107.038661. PMID 17942804.
- ↑ "Beyond Lipid-Lowering: Effects of Statins on Cardiovascular and Cerebrovascular Diseases and Cancer". Pharmaceuticals 15 (2): 151. January 2022. doi:10.3390/ph15020151. PMID 35215263.
- ↑ "A Review of Interleukin-1 in Heart Disease: Where Do We Stand Today?". Cardiology and Therapy 7 (1): 25–44. June 2018. doi:10.1007/s40119-018-0104-3. PMID 29417406.
- ↑ "Inflammatory markers in depression: A meta-analysis of mean differences and variability in 5,166 patients and 5,083 controls". Brain, Behavior, and Immunity 87: 901–909. July 2020. doi:10.1016/j.bbi.2020.02.010. PMID 32113908.
- ↑ "Vitamin A deficiency increases inflammatory responses". Scandinavian Journal of Immunology 44 (6): 578–84. December 1996. doi:10.1046/j.1365-3083.1996.d01-351.x. PMID 8972739.
- ↑ "Cocaine, not morphine, causes the generation of reactive oxygen species and activation of NF-kappaB in transiently cotransfected heart cells". Cardiovascular Toxicology 3 (2): 141–51. 2003. doi:10.1385/CT:3:2:141. PMID 14501032.
- ↑ "Role of reactive oxygen species, glutathione and NF-kappaB in apoptosis induced by 3,4-methylenedioxymethamphetamine ("Ecstasy") on hepatic stellate cells". Biochemical Pharmacology 67 (6): 1025–33. March 2004. doi:10.1016/j.bcp.2003.10.020. PMID 15006539.
- ↑ "Interaction of tumor cells with the microenvironment". Cell Communication and Signaling 9: 18. September 2011. doi:10.1186/1478-811X-9-18. PMID 21914164.
- ↑ 41.0 41.1 41.2 41.3 41.4 "Inflammation and cancer". Nature 420 (6917): 860–7. 2002. doi:10.1038/nature01322. PMID 12490959. Bibcode: 2002Natur.420..860C.
- ↑ "Opposing roles for complement component C5a in tumor progression and the tumor microenvironment". Journal of Immunology 189 (6): 2985–94. September 2012. doi:10.4049/jimmunol.1200846. PMID 22914051.
- ↑ "Sex steroid receptors in skeletal differentiation and epithelial neoplasia: is tissue-specific intervention possible?". BioEssays 31 (6): 629–41. June 2009. doi:10.1002/bies.200800138. PMID 19382224.
- ↑ "Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability". Carcinogenesis 30 (7): 1073–81. July 2009. doi:10.1093/carcin/bgp127. PMID 19468060.
- ↑ 45.0 45.1 45.2 45.3 45.4 45.5 45.6 45.7 "Inflammation-associated cancer development in digestive organs: mechanisms and roles for genetic and epigenetic modulation". Gastroenterology 143 (3): 550–563. September 2012. doi:10.1053/j.gastro.2012.07.009. PMID 22796521. http://repository.kulib.kyoto-u.ac.jp/dspace/bitstream/2433/160134/1/j.gastro.2012.07.009.pdf. Retrieved 9 June 2018.
- ↑ "Cancer-related inflammation". Nature 454 (7203): 436–44. July 2008. doi:10.1038/nature07205. PMID 18650914. Bibcode: 2008Natur.454..436M. https://air.unimi.it/bitstream/2434/145688/2/Cancer-related%20inflammation_Nature.pdf. Retrieved 9 June 2018.
- ↑ "Mediators of inflammation". Annual Review of Immunology 1: 335–59. 1983. doi:10.1146/annurev.iy.01.040183.002003. PMID 6399978.
- ↑ 48.0 48.1 48.2 48.3 "Chronic inflammation and cancer". Oncology 16 (2): 217–26, 229; discussion 230–2. February 2002. PMID 11866137.
- ↑ "Occurrence, Biological Consequences, and Human Health Relevance of Oxidative Stress-Induced DNA Damage". Chemical Research in Toxicology 29 (12): 2008–2039. December 2016. doi:10.1021/acs.chemrestox.6b00265. PMID 27989142.
- ↑ "Oxidatively induced DNA damage: mechanisms, repair and disease". Cancer Letters 327 (1–2): 26–47. December 2012. doi:10.1016/j.canlet.2012.01.016. PMID 22293091. Bibcode: 2012CancL.327...26D.
- ↑ "Oxidative stress and epigenetic instability in human hepatocarcinogenesis". Digestive Diseases 31 (5–6): 447–53. 2013. doi:10.1159/000355243. PMID 24281019.
- ↑ 52.0 52.1 "The emerging role of epigenetic modifiers in repair of DNA damage associated with chronic inflammatory diseases". Mutation Research 780: 69–81. 2017. doi:10.1016/j.mrrev.2017.09.005. PMID 31395351.
- ↑ "Nitrative and oxidative DNA damage in infection-related carcinogenesis in relation to cancer stem cells". Genes and Environment 38 (1). 2016. doi:10.1186/s41021-016-0055-7. PMID 28050219. Bibcode: 2016GeneE..38...26K.
- ↑ "Oxidative damage targets complexes containing DNA methyltransferases, SIRT1, and polycomb members to promoter CpG Islands". Cancer Cell 20 (5): 606–19. November 2011. doi:10.1016/j.ccr.2011.09.012. PMID 22094255.
- ↑ "Mismatch Repair Proteins Initiate Epigenetic Alterations during Inflammation-Driven Tumorigenesis". Cancer Research 77 (13): 3467–3478. July 2017. doi:10.1158/0008-5472.CAN-17-0056. PMID 28522752.
- ↑ "Genetic and epigenetic alterations in normal tissues have differential impacts on cancer risk among tissues". Proceedings of the National Academy of Sciences of the United States of America 115 (6): 1328–1333. February 2018. doi:10.1073/pnas.1717340115. PMID 29358395. Bibcode: 2018PNAS..115.1328Y.
- ↑ "Oxidative DNA damage as a potential early biomarker of Helicobacter pylori associated carcinogenesis". Pathology & Oncology Research 20 (4): 839–46. October 2014. doi:10.1007/s12253-014-9762-1. PMID 24664859.
- ↑ 58.0 58.1 "HIV infection, inflammation, immunosenescence, and aging". Annual Review of Medicine 62: 141–55. 2011-01-01. doi:10.1146/annurev-med-042909-093756. PMID 21090961.
- ↑ "Immune activation and HIV persistence: implications for curative approaches to HIV infection". Immunological Reviews 254 (1): 326–42. July 2013. doi:10.1111/imr.12065. PMID 23772629.
- ↑ "Increased immune activation precedes the inflection point of CD4 T cells and the increased serum virus load in human immunodeficiency virus infection". The Journal of Infectious Diseases 178 (2): 423–30. August 1998. doi:10.1086/515629. PMID 9697722.
- ↑ "The paradox of the immune response in HIV infection: when inflammation becomes harmful". Clinica Chimica Acta; International Journal of Clinical Chemistry 416: 96–9. February 2013. doi:10.1016/j.cca.2012.11.025. PMID 23228847.
- ↑ "Persistent inflammation in HIV infection: established concepts, new perspectives". Immunology Letters 161 (2): 184–8. October 2014. doi:10.1016/j.imlet.2014.01.008. PMID 24487059.
- ↑ "Lack of clinical AIDS in SIV-infected sooty mangabeys with significant CD4+ T cell loss is associated with double-negative T cells". The Journal of Clinical Investigation 121 (3): 1102–10. March 2011. doi:10.1172/JCI44876. PMID 21317533.
- ↑ "Simian immunodeficiency virus replicates to high levels in sooty mangabeys without inducing disease". Journal of Virology 72 (5): 3872–86. May 1998. doi:10.1128/JVI.72.5.3872-3886.1998. PMID 9557672.
- ↑ "Natural SIV hosts: showing AIDS the door". Science 335 (6073): 1188–93. March 2012. doi:10.1126/science.1217550. PMID 22403383. Bibcode: 2012Sci...335.1188C.
- ↑ 66.0 66.1 "Cell death by pyroptosis drives CD4 T-cell depletion in HIV-1 infection". Nature 505 (7484): 509–14. January 2014. doi:10.1038/nature12940. PMID 24356306. Bibcode: 2014Natur.505..509D.
- ↑ "IFI16 DNA sensor is required for death of lymphoid CD4 T cells abortively infected with HIV". Science 343 (6169): 428–32. January 2014. doi:10.1126/science.1243640. PMID 24356113. Bibcode: 2014Sci...343..428M.
- ↑ "Cell-to-Cell Transmission of HIV-1 Is Required to Trigger Pyroptotic Death of Lymphoid-Tissue-Derived CD4 T Cells". Cell Reports 12 (10): 1555–1563. September 2015. doi:10.1016/j.celrep.2015.08.011. PMID 26321639.
- ↑ "Dissecting How CD4 T Cells Are Lost During HIV Infection". Cell Host & Microbe 19 (3): 280–91. March 2016. doi:10.1016/j.chom.2016.02.012. PMID 26962940.
- ↑ Study of VX-765 in Subjects With Treatment-resistant Partial Epilepsy – Full Text View – ClinicalTrials.gov. 19 December 2013. https://clinicaltrials.gov/ct2/show/NCT01048255. Retrieved 2016-05-21.
- ↑ "So depression is an inflammatory disease, but where does the inflammation come from?". BMC Medicine 11. September 2013. doi:10.1186/1741-7015-11-200. PMID 24228900.
- ↑ "Stereotypes, Prejudice, and Depression: The Integrated Perspective". Perspectives on Psychological Science 7 (5): 427–49. September 2012. doi:10.1177/1745691612455204. PMID 26168502.
- ↑ "Inflammation: depression fans the flames and feasts on the heat". The American Journal of Psychiatry 172 (11): 1075–91. November 2015. doi:10.1176/appi.ajp.2015.15020152. PMID 26357876.
- ↑ "Is depression a kind of allergic reaction?". The Guardian. 2015-01-04. https://www.theguardian.com/lifeandstyle/2015/jan/04/depression-allergic-reaction-inflammation-immune-system.
- ↑ "Comparison of cytokine levels in depressed, manic and euthymic patients with bipolar disorder". Journal of Affective Disorders 116 (3): 214–7. August 2009. doi:10.1016/j.jad.2008.12.001. PMID 19251324.
- ↑ "The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine". Molecular Psychiatry 11 (7): 680–4. July 2006. doi:10.1038/sj.mp.4001805. PMID 16491133.
- ↑ "Reconceptualizing major depressive disorder as an infectious disease". Biology of Mood & Anxiety Disorders 4. 2014. doi:10.1186/2045-5380-4-10. PMID 25364500.
- ↑ "Inflammatory and blood gas markers of COVID-19 delirium compared to non-COVID-19 delirium: a cross-sectional study". Aging & Mental Health 26 (10): 2054–2061. October 2022. doi:10.1080/13607863.2021.1989375. PMID 34651536. https://discovery.ucl.ac.uk/id/eprint/10136824/. Retrieved 20 February 2023.
- ↑ "Physiology, Vasodilation". StatPearls. Treasure Island (FL): StatPearls Publishing. 2021. https://www.ncbi.nlm.nih.gov/books/NBK557562/. Retrieved 22 September 2021.
- ↑ 80.0 80.1 "Secret talk between adipose tissue and central nervous system via secreted factors-an emerging frontier in the neurodegenerative research". Journal of Neuroinflammation 13 (1). March 2016. doi:10.1186/s12974-016-0530-x. PMID 27012931.
- ↑ "Adipose tissue as an endocrine organ". The Journal of Clinical Endocrinology and Metabolism 89 (6): 2548–56. June 2004. doi:10.1210/jc.2004-0395. PMID 15181022.
- ↑ "Elevated levels of interleukin 6 are reduced in serum and subcutaneous adipose tissue of obese women after weight loss". The Journal of Clinical Endocrinology and Metabolism 85 (9): 3338–42. September 2000. doi:10.1210/jcem.85.9.6839. PMID 10999830.
- ↑ "beta-Adrenergic regulation of IL-6 release from adipose tissue: in vivo and in vitro studies". The Journal of Clinical Endocrinology and Metabolism 86 (12): 5864–9. December 2001. doi:10.1210/jcem.86.12.8104. PMID 11739453.
- ↑ 84.0 84.1 84.2 84.3 84.4 84.5 84.6 "Leptin regulates proinflammatory immune responses". FASEB Journal 12 (1): 57–65. January 1998. doi:10.1096/fasebj.12.1.57. PMID 9438411.
- ↑ 85.0 85.1 85.2 85.3 85.4 85.5 85.6 "Inflammatory cytokine concentrations are acutely increased by hyperglycemia in humans: role of oxidative stress". Circulation 106 (16): 2067–72. October 2002. doi:10.1161/01.CIR.0000034509.14906.AE. PMID 12379575.
- ↑ "The anti-inflammatory effect of exercise". Journal of Applied Physiology 98 (4): 1154–62. April 2005. doi:10.1152/japplphysiol.00164.2004. PMID 15772055. Bibcode: 2005JAPh...98.1154P.
- ↑ "Waist circumference as the predominant contributor to the micro-inflammatory response in the metabolic syndrome: a cross sectional study". Journal of Inflammation 7: 35. July 2010. doi:10.1186/1476-9255-7-35. PMID 20659330.
- ↑ "Therapeutic approaches targeting inflammation for diabetes and associated cardiovascular risk". The Journal of Clinical Investigation 127 (1): 83–93. January 2017. doi:10.1172/jci88884. PMID 28045401.
- ↑ "Obesity and C-reactive protein in various populations: a systematic review and meta-analysis". Obesity Reviews 14 (3): 232–44. March 2013. doi:10.1111/obr.12003. PMID 23171381.
- ↑ "Inflammation in wound repair: molecular and cellular mechanisms". The Journal of Investigative Dermatology 127 (3): 514–25. March 2007. doi:10.1038/sj.jid.5700701. PMID 17299434.
- ↑ "Mice lacking Smad3 show accelerated wound healing and an impaired local inflammatory response". Nature Cell Biology 1 (5): 260–6. September 1999. doi:10.1038/12971. PMID 10559937.
- ↑ "Bidirectional regulation of macrophage function by TGF-beta". Microbes and Infection 1 (15): 1275–82. December 1999. doi:10.1016/S1286-4579(99)00257-9. PMID 10611755. https://zenodo.org/record/1260212. Retrieved 11 September 2019.
- ↑ "Transforming growth factor-beta 1 inhibition of macrophage activation is mediated via Smad3". The Journal of Biological Chemistry 275 (47): 36653–8. November 2000. doi:10.1074/jbc.M004536200. PMID 10973958.
- ↑ "Regulatory role of endogenous interleukin-10 in cutaneous inflammatory response of murine wound healing". Biochemical and Biophysical Research Communications 265 (1): 194–9. November 1999. doi:10.1006/bbrc.1999.1455. PMID 10548513. Bibcode: 1999BBRC..265..194S.
- ↑ "Controlling the resolution of acute inflammation: a new genus of dual anti-inflammatory and proresolving mediators". Journal of Periodontology 79 (8 Suppl): 1520–6. August 2008. doi:10.1902/jop.2008.080231. PMID 18673006.
- ↑ "The resolution of inflammation: Principles and challenges". Seminars in Immunology 27 (3): 149–60. May 2015. doi:10.1016/j.smim.2015.03.014. PMID 25911383.
- ↑ "The role of apoptosis in wound healing". The International Journal of Biochemistry & Cell Biology 30 (9): 1019–30. September 1998. doi:10.1016/S1357-2725(98)00058-2. PMID 9785465.
- ↑ "Regulation of lung injury and repair by Toll-like receptors and hyaluronan". Nature Medicine 11 (11): 1173–9. November 2005. doi:10.1038/nm1315. PMID 16244651.
- ↑ "Resolution of lung inflammation by CD44". Science 296 (5565): 155–8. April 2002. doi:10.1126/science.1069659. PMID 11935029. Bibcode: 2002Sci...296..155T.
- ↑ "Inflammation dampened by gelatinase A cleavage of monocyte chemoattractant protein-3". Science 289 (5482): 1202–6. August 2000. doi:10.1126/science.289.5482.1202. PMID 10947989. Bibcode: 2000Sci...289.1202M.
- ↑ "Resolution of inflammation: the beginning programs the end". Nature Immunology 6 (12): 1191–7. December 2005. doi:10.1038/ni1276. PMID 16369558.
External links
- Inflammation at the US National Library of Medicine Medical Subject Headings (MeSH)
 |  |
 EncycloReader
is supported by the
EncycloReader
is supported by the