Coot
Topic: Software
 From HandWiki - Reading time: 5 min
From HandWiki - Reading time: 5 min
Coots are medium-sized water birds that are members of the rail family, Rallidae. They constitute the genus Fulica, the name being the Latin term for "coot". Coots have predominantly black plumage, and—unlike many rails—they are usually easy to see, often swimming in open water.
Taxonomy and systematics
The genus Fulica was introduced in 1758 by the Swedish naturalist Carl Linnaeus in the tenth edition of his Systema Naturae.[1] The genus name is the Latin word for a Eurasian coot.[2] The name was used by the Swiss naturalist Conrad Gessner in 1555.[3] The type species is the Eurasian coot.[4]
A group of coots is referred to as a covert[5] or cover.[6]
Species
The genus contains 10 extant species and one which is now extinct.[7]
| Image | Scientific name | Common name | Distribution |
|---|---|---|---|
| 120px | Fulica alai Peale, 1848 | Hawaiian coot or ʻAlae keʻokeʻo | Hawaii |
| 120px | Fulica americana Gmelin, 1789 | American coot | southern Quebec to the Pacific coast of North America and as far south as northern South America |
| 120px | Fulica ardesiaca Tschudi, 1843 | Andean coot | Argentina, Bolivia, Chile, Colombia, Ecuador, Peru |
| 120px | Fulica armillata Vieillot, 1817 | red-gartered coot | Argentina, southern Brazil, Chile, Paraguay, Uruguay |
| 120px | Fulica atra Linnaeus, 1758 | Eurasian coot or common coot | Europe, Asia, Australia, and Africa |
| 120px | Fulica cornuta Bonaparte, 1853 | horned coot | Argentina, Bolivia, Chile |
| 120px | Fulica cristata Gmelin, 1789 | red-knobbed coot | Africa, Iberian Peninsula |
| 120px | Fulica gigantea Eydoux & Souleyet, 1841 | giant coot | Argentina, Bolivia, Chile, Peru |
| 120px | Fulica leucoptera Vieillot, 1817 | white-winged coot | Argentina, Bolivia, Brazil, Chile, Falkland Islands, Paraguay, Uruguay |
| 120px | Fulica rufifrons Philppi & Landbeck, 1861 | red-fronted coot | Argentina, southern Brazil, Chile, Paraguay, southern Peru, Uruguay |
Extinct species
Recently extinct species
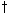 Fulica newtonii Milne-Edwards, 1867 – Mascarene coot (extinct, c. 1700)
Fulica newtonii Milne-Edwards, 1867 – Mascarene coot (extinct, c. 1700)
Late Quaternary species
- Fulica chathamensis Forbes, 1892 – Chatham Island coot (early Holocene of the Chatham Islands)
- Fulica montanei Alarcón-Muñoz, Labarca & Soto-Acuña, 2020 (late Pleistocene to early Holocene of Chile)[8]
- Fulica prisca Hamilton, 1893 – New Zealand coot (early Holocene of New Zealand)
- Fulica shufeldti (Brodkorb, 1964) (late Pleistocene of Florida) possibly a paleosubspecies of Fulica americana; formerly F. minor[9]
Fossil species
- Fulica infelix Brodkorb, 1961 – (early Pliocene of Juntura, Malheur County, Oregon, USA)
Description
Coots have prominent frontal shields or decoration on their foreheads, with red to dark red eyes and coloured bills. Many have white on the under tail. The featherless shield gave rise to the expression "as bald as a coot",[10] which the Oxford English Dictionary cites in use as early as 1430. Coots have long toes with broad lobes of skin that allow them to kick and propel themselves through the water. The lobes of skin fold back each time the coot lifts its foot, allowing them to walk on dry land while also providing support in mucky terrain.[11] They tend to have short, rounded wings and are weak fliers, though northern species nevertheless can cover long distances. They typically congregate in large rafts in open water. Along these rafts coots may lay eggs in their own nest or in some other bird's. Depending on the species of coot the eggs can vary in color: buff, pinkish buff or buff-gray speckled with dark brown, purplish brown, or black.[12]
Distribution and habitat
The greatest species variety occurs in South America, and the genus likely originated there. They are common in Europe and North America.[13] Coot species that migrate do so at night. The American coot has been observed rarely in Britain and Ireland, while the Eurasian coot is found across Asia, Australia and parts of Africa. In southern Louisiana, the coot is referred to by the French name "poule d'eau", which translates into English as "water hen".[14]
Behaviour and ecology
Coots are omnivorous, eating mainly plant material, but also small animals, fish and eggs.[15] They are aggressively territorial during the breeding season, but are otherwise often found in sizeable flocks on the shallow vegetated lakes they prefer.
Chick mortality occurs mainly due to starvation rather than predation as coots have difficulty feeding a large family of hatchlings on the tiny shrimp and insects that they collect. Many chicks die in the first 10 days after hatching, when they are most dependent on adults for food.[16] Coots can be very brutal to their own young under pressure such as the lack of food, and after about three days they start attacking their own chicks when they beg for food. After a short while, these attacks concentrate on the weaker chicks, who eventually give up begging and die. The coot may eventually raise only two or three out of nine hatchlings.[17] In this attacking behaviour, the parents are said to "tousle" their young. This can result in the death of the chick.[18]
References
- ↑ Linnaeus, Carl (1758) (in Latin). Systema Naturae per regna tria naturae, secundum classes, ordines, genera, species, cum characteribus, differentiis, synonymis, locis. 1 (10th ed.). Holmiae (Stockholm): Laurentii Salvii. p. 152. https://www.biodiversitylibrary.org/page/727059.
- ↑ Jobling, James A. (2010). The Helm Dictionary of Scientific Bird Names. London: Christopher Helm. p. 165. ISBN 978-1-4081-2501-4.
- ↑ Gesner, Conrad (1555) (in Latin). Historiae animalium liber III qui est de auium natura. Adiecti sunt ab initio indices alphabetici decem super nominibus auium in totidem linguis diuersis: & ante illos enumeratio auium eo ordiné quo in hoc volumine continentur. Zurich: Froschauer. p. 375. https://www.biodiversitylibrary.org/page/52661257.
- ↑ Peters, James Lee, ed (1934). Check-List of Birds of the World. 2. Cambridge, Massachusetts: Harvard University Press. p. 211. https://www.biodiversitylibrary.org/page/14483024.
- ↑ "What do you call a group of ...?". Oxford Dictionaries. Oxford University Press. http://www.oxforddictionaries.com/page/collectivenouns_us.
- ↑ "Baltimore Bird Club. Group Name for Birds: A Partial List". http://baltimorebirdclub.org/gnlist.html.
- ↑ Gill, Frank; Donsker, David; Rasmussen, Pamela, eds (July 2021). "Flufftails, finfoots, rails, trumpeters, cranes, limpkin". IOC World Bird List Version 11.2. International Ornithologists' Union. https://www.worldbirdnames.org/bow/flufftails/.
- ↑ Alarcón-Muñoz, Jhonatan; Labarca, Rafael; Soto-Acuña, Sergio (2020-12-01). "The late Pleistocene–early Holocene rails (Gruiformes: Rallidae) of Laguna de Tagua Tagua Formation, central Chile, with the description of a new extinct giant coot" (in en). Journal of South American Earth Sciences 104. doi:10.1016/j.jsames.2020.102839. Bibcode: 2020JSAES.10402839A. http://www.sciencedirect.com/science/article/pii/S0895981120303825.
- ↑ Jehl, Joseph R. (1967). "Pleistocene Birds from Fossil Lake, Oregon". The Condor 69 (1): 24–27. doi:10.2307/1366369. http://sora.unm.edu/node/101598.
- ↑ "Coot | The Wildlife Trusts" (in en). https://www.wildlifetrusts.org/wildlife-explorer/birds/wading-birds/coot.
- ↑ "American Coot Overview, All About Birds, Cornell Lab of Ornithology" (in en). https://www.allaboutbirds.org/guide/American_Coot/overview.
- ↑ "American Coot Life History, All About Birds, Cornell Lab of Ornithology" (in en). https://www.allaboutbirds.org/guide/American_Coot/lifehistory.
- ↑ Olson, Storrs L. (1974). "The Pleistocene Rails of North America." Museum of Natural History.
- ↑ "American Coot". http://losbird.org/labirds/amco.htm.
- ↑ Ornithology, British Trust for (2015-04-07). "Coot" (in en). https://www.bto.org/understanding-birds/birdfacts/coot.
- ↑ "This Coot has a Secret! - NatureOutside". 20 June 2015. http://www.natureoutside.com/this-coot-has-a-secret/.
- ↑ The Life of Birds, David Attenborough. The Problems of Parenthood. 10:20.
- ↑ Clutton-Brock, TH., The Evolution of Parental Care, Princeton University Press, 1991 p. 203.
External links
* Coot videos on the Internet Bird Collection
 Beach, Chandler B., ed (1914). "Coot". The New Student's Reference Work. Chicago: F. E. Compton and Co.
Beach, Chandler B., ed (1914). "Coot". The New Student's Reference Work. Chicago: F. E. Compton and Co.
Template:Gruiformes Wikidata ☰ Q637533 entry
 |  |
 EncycloReader
is supported by the
EncycloReader
is supported by the