Acute intermittent porphyria
 From Mdwiki - Reading time: 11 min
From Mdwiki - Reading time: 11 min| Acute intermittent porphyria | |
|---|---|
| Other names: HMBS deficiency,[1] Swedish porphyria, pyrroloporphyria, intermittent acute porphyria | |
 | |
| Porphobilinogen | |
| Specialty | Medical genetics |
Acute intermittent porphyria (AIP) is a rare metabolic disorder affecting the production of heme resulting from a deficiency of the porphobilinogen deaminase. It is the most common of the acute porphyrias.[2][3][4]
Signs and symptoms[edit | edit source]
The clinical presentation of AIP is highly variable and non-specific. The patients are typically asymptomatic, with most gene carriers having no family history because the condition had remained latent for several generations. The syndrome marked by acute attacks affects only 10% of gene carriers.[5] The mean age at diagnosis is 33 years old.[6] Like other porphyrias, AIP is more likely to present in women.[7] A distinguishing feature of AIP that separates it from other porphyrias is the absence of photosensitive cutaneous symptoms that occur in addition to acute attacks.[8]
Acute attacks[edit | edit source]
AIP is one of the four porphyrias that presents as an acute attack. 90% of affected individuals never experience an acute attack and are asymptomatic, while an estimated 5% of affected individuals experience repeat attacks.[9] Attacks are most common in young adult women and are rare before puberty or after menopause.[10] Severe acute attacks may require hospitalization. Patients usually experience symptoms in attacks that last from several hours to a few days. Between attacks, patients are asymptomatic.[citation needed]
The most frequent presenting symptoms are abdominal pain and tachycardia.[11] The abdominal pain is typically severe, colicky, poorly localized, and often associated with pain in back and legs.[12][13] Patients may also present with vomiting and constipation, but diarrhea is unusual.[14] The onset of back and leg pain is characterized by severe pain and stiffness in back and thighs followed by loss of tendon reflexes and paralysis.[15] Psychiatric symptoms are present, such as paranoid schizophrenia-like features but rarely psychosis and hallucinations.[16] The acute attacks classically present with dark-red photosensitive urine (often called port-wine urine), but this is a nonspecific symptom.[17] Physical examination often shows no abnormalities.[18]
Hyponatremia is the most common electrolyte abnormality during acute attacks, occurring in 40% of patients and presenting as SIADH.[19] Hypomagnesemia is also common. There are no pathognomonic signs or symptoms.[citation needed]
The most common identified triggers for acute attacks are medications, weight loss diets, and surgery.[20] Many medications have been associated with AIP including antibiotics, hormonal contraceptives, seizure medications, anesthetics, and HIV treatment drugs.[21]
Pathophysiology[edit | edit source]

Porphyrias are caused by mutations in genes that encode enzymes in heme synthesis. In normal physiology, heme synthesis begins in the mitochondrion, proceeds into the cytoplasm, and finishes back in the mitochondrion. Heme is produced in all cells, but 80% of all heme is produced in erythropoietic cells in bone marrow and 15% in parenchymal cells in the liver, where turnover of hemoproteins is high. In AIP, over 100 mutations have been identified on the long arm of chromosome 11 at the HMBS gene, which codes for the cytoplasmic enzyme porphobilinogen deaminase.[23]
This deficiency prevents heme synthesis, which can not be completed and the metabolite porphobilinogen accumulates in the cytoplasm.[24]
AIP is an autosomal dominant porphyria resulting in about 50% normal activity of the affected enzyme.[25]
The penetrance of AIP is incomplete with only 10% of gene carriers experiencing acute attacks suggesting role for other modifying genes or environment.[26][27][28]
The exact mechanism of acute attacks is not clear. The most favored hypothesis is that porpholobilinogen buildup causes a toxic effects on neurons. The autonomic and peripheral nervous system are more vulnerable than the central nervous system because they are not protected by the blood-brain barrier.[29] This explains findings such as abdominal pain and tachycardia. Some individuals may be more likely to develop paresis based on increased susceptibility of neurons to toxins.[30]
Diagnosis[edit | edit source]
-
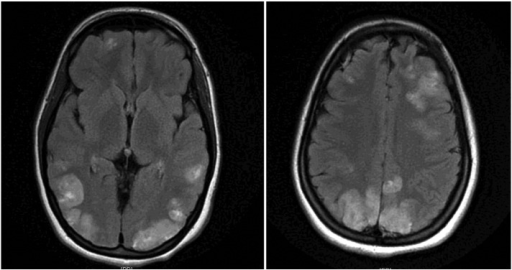
Image shows faint multifocal and bilateral hyperintensities in the frontal lobes and parietooccipital lobes, extending to the left greater than right posterior temporal regions
-

Acute intermittent porphyria-High coloured urine turned reddish brown on standing
.png)

The initial diagnosis of acute porphyria is confirmed by urinalysis, including the common method, the Watson-Schwartz test. Elevated urine porphobilinogen confirms diagnosis of AIP, hereditary coproporphyria (HCP), or variegate porphyria (VP). A positive test should be indicated with an increase of five times normal, not just a slight increase which can occur with dehydration. To distinguish between AIP from HCP and VP, fecal porphyrin levels are normal in AIP but elevated in HCP and VP.[citation needed]
Rapid, accurate diagnosis is important. Delays in diagnosis may result in permanent neurological damage or death.[citation needed]
Treatment[edit | edit source]

If drugs have caused the attack, discontinuing the offending substances is essential. A high-carbohydrate (10% glucose) infusion is recommended, which may aid in recovery.[citation needed]
Hematin and heme arginate is the treatment of choice during an acute attack. Heme is not a curative treatment, but can shorten attacks and reduce the intensity of an attack. Side-effects are rare but can be serious.[citation needed] Pain is extremely severe and almost always requires the use of opiates to reduce it to tolerable levels. Pain should be treated as early as medically possible due to its severity.
Nausea can be severe; it may respond to phenothiazine drugs but is sometimes intractable. Hot water baths or showers may lessen nausea temporarily, but can present a risk of burns or falls.[31]
Seizures often accompany this disease. Most seizure medications exacerbate this condition due to their induction of cytochrome P450. Treatment can be problematic: Barbiturates and primidone must be avoided as they commonly precipitate symptoms.[32] Some benzodiazepines are safe, and, when used in conjunction with newer anti-seizure medications such as gabapentin, offer a possible regimen for seizure control.[citation needed]
Prevalence[edit | edit source]
In terms of prevalence of Acute hepatic porphyria we find that Acute intermediate porphyria is the most prevalent/frequent.[33]
Society and culture[edit | edit source]
One of the many hypothesized diagnoses of the artist Vincent van Gogh is that he and his siblings, in particular his brother Theo, suffered from AIP and syphilis.[34] Another theorized sufferer was King George III of the United Kingdom[35] who even had a medallion struck to commemorate his "curing". His descendant Prince William of Gloucester was reliably diagnosed with variegate porphyria in 1968.[36] It is probable that the philosopher Jean-Jacques Rousseau suffered from porphyria.[37][38][39][40] It has even been suggested that Vlad III, Prince of Wallachia, more commonly known by his surname Dracula, suffered from porphyria.
References[edit | edit source]
- ↑ "Acute intermittent porphyria (Concept Id: C0162565) - MedGen - NCBI". www.ncbi.nlm.nih.gov.
- ↑ Whatley SD, Roberts AG, Llewellyn DH, Bennett CP, Garrett C, Elder GH (September 2000). "Non-erythroid form of acute intermittent porphyria caused by promoter and frameshift mutations distant from the coding sequence of exon 1 of the HMBS gene". Human Genetics. 107 (3): 243–8. doi:10.1007/s004390000356. PMID 11071386.
- ↑ Solis C, Martinez-Bermejo A, Naidich TP, Kaufmann WE, Astrin KH, Bishop DF, Desnick RJ (November 2004). "Acute intermittent porphyria: studies of the severe homozygous dominant disease provides insights into the neurologic attacks in acute porphyrias". Archives of Neurology. 61 (11): 1764–70. doi:10.1001/archneur.61.11.1764. PMID 15534187.
- ↑ Diseases of Tetrapyrrole Metabolism - Refsum Disease and the Hepatic Porphyrias at eMedicine
- ↑ Narang, Neatu; Banerjee, A; Kotwal, J; Kaur, Jasmeet; Sharma, YV; Sharma, CS (April 2003). "Psychiatric Manifestations in three cases of Acute Intermittent Porphyria". Medical Journal, Armed Forces India. 59 (2): 171–173. doi:10.1016/S0377-1237(03)80075-8. ISSN 0377-1237. PMC 4923792. PMID 27407502.
- ↑ Elder, George; Harper, Pauline; Badminton, Michael; Sandberg, Sverre; Deybach, Jean-Charles (September 2013). "The incidence of inherited porphyrias in Europe". Journal of Inherited Metabolic Disease. 36 (5): 849–857. doi:10.1007/s10545-012-9544-4. ISSN 1573-2665. PMID 23114748.
- ↑ Puy, Hervé; Gouya, Laurent; Deybach, Jean-Charles (2010-03-13). "Porphyrias". Lancet. 375 (9718): 924–937. doi:10.1016/S0140-6736(09)61925-5. ISSN 1474-547X. PMID 20226990.
- ↑ Stein, Penelope; Badminton, Mike; Barth, Julian; Rees, David; Stewart, M. Felicity; British and Irish Porphyria Network (May 2013). "Best practice guidelines on clinical management of acute attacks of porphyria and their complications". Annals of Clinical Biochemistry. 50 (Pt 3): 217–223. doi:10.1177/0004563212474555. ISSN 1758-1001. PMID 23605132.
- ↑ Elder, George; Harper, Pauline; Badminton, Michael; Sandberg, Sverre; Deybach, Jean-Charles (2012-11-01). "The incidence of inherited porphyrias in Europe". Journal of Inherited Metabolic Disease. 36 (5): 849–857. doi:10.1007/s10545-012-9544-4. ISSN 0141-8955. PMID 23114748.
- ↑ Besur, Siddesh; Hou, Wehong; Schmeltzer, Paul; Bonkovsky, Herbert L. (2014-11-03). "Clinically important features of porphyrin and heme metabolism and the porphyrias". Metabolites. 4 (4): 977–1006. doi:10.3390/metabo4040977. ISSN 2218-1989. PMC 4279155. PMID 25372274.
- ↑ Besur, Siddesh; Schmeltzer, Paul; Bonkovsky, Herbert L. (September 2015). "Acute Porphyrias". The Journal of Emergency Medicine. 49 (3): 305–312. doi:10.1016/j.jemermed.2015.04.034. ISSN 0736-4679. PMID 26159905.
- ↑ Besur, Siddesh; Schmeltzer, Paul; Bonkovsky, Herbert L. (September 2015). "Acute Porphyrias". The Journal of Emergency Medicine. 49 (3): 305–312. doi:10.1016/j.jemermed.2015.04.034. ISSN 0736-4679. PMID 26159905.
- ↑ Kauppinen, Raili (15–21 Jan 2005). "Porphyrias". Lancet. 365 (9455): 241–252. doi:10.1016/s0140-6736(05)70154-9. ISSN 1474-547X. PMID 15652607.
- ↑ Kauppinen, Raili (15–21 Jan 2005). "Porphyrias". Lancet. 365 (9455): 241–252. doi:10.1016/s0140-6736(05)70154-9. ISSN 1474-547X. PMID 15652607.
- ↑ Pischik, E.; Kauppinen, R. (2009-02-16). "Neurological manifestations of acute intermittent porphyria". Cellular and Molecular Biology (Noisy-Le-Grand, France). 55 (1): 72–83. ISSN 1165-158X. PMID 19268005.
- ↑ Narang, Neatu; Banerjee, A; Kotwal, J; Kaur, Jasmeet; Sharma, YV; Sharma, CS (April 2003). "Psychiatric Manifestations in three cases of Acute Intermittent Porphyria". Medical Journal, Armed Forces India. 59 (2): 171–173. doi:10.1016/S0377-1237(03)80075-8. ISSN 0377-1237. PMC 4923792. PMID 27407502.
- ↑ Yuan, Tao; Li, Yu-Hui; Wang, Xi; Gong, Feng-Ying; Wu, Xue-Yan; Fu, Yong; Zhao, Wei-Gang (2015-07-20). "Acute Intermittent Porphyria: A Diagnostic Challenge for Endocrinologist". Chinese Medical Journal. 128 (14): 1980–1981. doi:10.4103/0366-6999.160621. ISSN 0366-6999. PMC 4717930. PMID 26168842.
- ↑ Karim, Zoubida; Lyoumi, Said; Nicolas, Gael; Deybach, Jean-Charles; Gouya, Laurent; Puy, Hervé (September 2015). "Porphyrias: A 2015 update". Clinics and Research in Hepatology and Gastroenterology. 39 (4): 412–425. doi:10.1016/j.clinre.2015.05.009. ISSN 2210-7401. PMID 26142871.
- ↑ Karim, Zoubida; Lyoumi, Said; Nicolas, Gael; Deybach, Jean-Charles; Gouya, Laurent; Puy, Hervé (September 2015). "Porphyrias: A 2015 update". Clinics and Research in Hepatology and Gastroenterology. 39 (4): 412–425. doi:10.1016/j.clinre.2015.05.009. ISSN 2210-741X. PMID 26142871.
- ↑ Bonkovsky, Herbert L.; Maddukuri, Vinaya C.; Yazici, Cemal; Anderson, Karl E.; Bissell, D. Montgomery; Bloomer, Joseph R.; Phillips, John D.; Naik, Hetanshi; Peter, Inga (December 2014). "Acute porphyrias in the USA: features of 108 subjects from porphyrias consortium". The American Journal of Medicine. 127 (12): 1233–1241. doi:10.1016/j.amjmed.2014.06.036. ISSN 1555-7162. PMC 4563803. PMID 25016127.
- ↑ Dhital, Rashmi; Basnet, Sijan; Poudel, Dilli Ram; Bhusal, Khema Raj (2017-06-06). "Acute intermittent porphyria: a test of clinical acumen". Journal of Community Hospital Internal Medicine Perspectives. 7 (2): 100–102. doi:10.1080/20009666.2017.1317535. ISSN 2000-9666. PMC 5473191. PMID 28638573.
- ↑ Ricci, Andrea; Di Pierro, Elena; Marcacci, Matteo; Ventura, Paolo (26 November 2021). "Mechanisms of Neuronal Damage in Acute Hepatic Porphyrias". Diagnostics (Basel, Switzerland). 11 (12): 2205. doi:10.3390/diagnostics11122205. ISSN 2075-4418.
- ↑ Herrick, Ariane L.; McColl, Kenneth E. L. (April 2005). "Acute intermittent porphyria". Best Practice & Research. Clinical Gastroenterology. 19 (2): 235–249. doi:10.1016/j.bpg.2004.10.006. ISSN 1521-6918. PMID 15833690.
- ↑ Kauppinen, R.; Mustajoki, S.; Pihlaja, H.; Peltonen, L.; Mustajoki, P. (1995). "Acute intermittent porphyria in Finland: 19 mutations in the porphobilinogen deaminase gene". Human Molecular Genetics. 4 (2): 215–222. doi:10.1093/hmg/4.2.215. ISSN 0964-6906. PMID 7757070.
- ↑ Whatley, Sharon D.; Badminton, Michael N. (1993), Adam, Margaret P.; Ardinger, Holly H.; Pagon, Roberta A.; Wallace, Stephanie E. (eds.), "Acute Intermittent Porphyria", GeneReviews®, University of Washington, Seattle, PMID 20301372, archived from the original on 2021-03-09, retrieved 2018-11-01
- ↑ Aarsand AK, Petersen PH, Sandberg S (April 2006). "Estimation and application of biological variation of urinary delta-aminolevulinic acid and porphobilinogen in healthy individuals and in patients with acute intermittent porphyria". Clinical Chemistry. 52 (4): 650–6. doi:10.1373/clinchem.2005.060772. PMID 16595824.
- ↑ Lannfelt L, Wetterberg L, Gellerfors P, Lilius L, Floderus Y, Thunell S (November 1989). "Mutations in acute intermittent porphyria detected by ELISA measurement of porphobilinogen deaminase". Journal of Clinical Chemistry and Clinical Biochemistry. 27 (11): 857–62. CiteSeerX 10.1.1.634.1622. doi:10.1515/cclm.1989.27.11.857. PMID 2607315.
- ↑ Pischik E, Kauppinen R (2015). "An update of clinical management of acute intermittent porphyria". The Application of Clinical Genetics. 8: 201–14. doi:10.2147/TACG.S48605. PMC 4562648. PMID 26366103.
- ↑ Laiwah, A C; Goldberg, A; Moore, M R (May 1983). "Pathogenesis and treatment of acute intermittent porphyria: discussion paper". Journal of the Royal Society of Medicine. 76 (5): 386–392. doi:10.1177/014107688307600512. ISSN 0141-0768. PMC 1439174. PMID 6864706.
- ↑ Chen, Brenden; Solis-Villa, Constanza; Hakenberg, Jörg; Qiao, Wanqiong; Srinivasan, Ramakrishnan R.; Yasuda, Makiko; Balwani, Manisha; Doheny, Dana; Peter, Inga (November 2016). "Acute Intermittent Porphyria: Predicted Pathogenicity of HMBS Variants Indicates Extremely Low Penetrance of the Autosomal Dominant Disease". Human Mutation. 37 (11): 1215–1222. doi:10.1002/humu.23067. ISSN 1059-7794. PMC 5063710. PMID 27539938.
- ↑ *American Porphyria Foundation. "About Porphyria: Acute Intermittent Porhyria" Archived April 25, 2008, at the Wayback Machine, 2007,
- ↑ Marcucci L (2004). PathCards. Baltimore, MD: Lippincott Willians & Wilkins. pp. 105–106. ISBN 978-0-7817-4399-0.
- ↑ "HMBS deficiency - WikiProjectMed". mdwiki.org.
- ↑ Arnold WN (1992). Vincent van Gogh : chemicals, crises, and creativity. Boston: Birkhäuser. ISBN 978-0-8176-3616-6.[page needed]
- ↑ Macalpine I, Hunter R (January 1966). "The "insanity" of King George 3d: a classic case of porphyria". British Medical Journal. 1 (5479): 65–71. doi:10.1136/bmj.1.5479.65. PMC 1843211. PMID 5323262.
- ↑ Röhl JC, Warren M, Hunt D (1998). Purple secret : genes, 'madness' and the royal houses of Europe. London: Corgi Books. ISBN 978-0-552-14550-3.[page needed]
- ↑ Bartolo A (1995). "Le maschere dell'io: Rousseau e la menzogna autobiografica" [The ego masks: Rousseau and the autobiographical lie]. Schena (in italiano): 113.
- ↑ "Jean-Jacques Rousseau l'errante" [Jean-Jacques Rousseau the wanderer]. La Letteratura e Noi - diretto da Romano Luperini (in italiano). Archived from the original on 19 November 2015. Retrieved 18 November 2015.
- ↑ Mejía-Rivera O (8 June 2012). "Las enfermedades de Jean-Jacques Rousseau" [The diseases of Jean-Jacques Rousseau]. Revista Aleph (in español). Archived from the original on 19 November 2015. Retrieved 18 November 2015.
- ↑ Androutsos G, Geroulanos S (December 2000). "[Acute intermittent porphyria: a new hypothesis to explain Jean-Jacques Rousseau's urinary disorders]". Progres en Urologie. 10 (6): 1282–9. PMID 11217576.
External links[edit | edit source]
| Classification | |
|---|---|
| External resources |
Original source: https://mdwiki.org/wiki/Acute intermittent porphyria
Status: article is cached
 EncycloReader
is supported by the
EncycloReader
is supported by the