Table of Contents
- 1 Medical uses
- 2 Contraindications
- 3 Side effects
- 4 Overdose
- 5 Interactions
- 6 Pharmacology
- 7 Chemistry
- 8 History
- 9 Society and culture
- 10 References
- 11 External links Categories
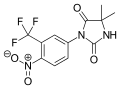
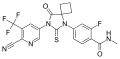
- 4-[7-[6-cyano-5-(trifluoromethyl)pyridin-3-yl]-8-oxo-6-sulfanylidene-5,7-diazaspiro[3.4]octan-5-yl]-2-fluoro-N-methylbenzamide
- CNC(=O)C1=C(C=C(C=C1)N2C(=S)N(C(=O)C23CCC3)C4=CN=C(C(=C4)C(F)(F)F)C#N)F
- InChI=1S/C21H15F4N5O2S/c1-27-17(31)13-4-3-11(8-15(13)22)30-19(33)29(18(32)20(30)5-2-6-20)12-7-14(21(23,24)25)16(9-26)28-10-12/h3-4,7-8,10H,2,5-6H2,1H3,(H,27,31)
- Key:HJBWBFZLDZWPHF-UHFFFAOYSA-N
-

-
-

-

-

-
Apalutamide
- ↑ 1.0 1.1 "Apalutamide Monograph for Professionals". Drugs.com. Archived from the original on 10 December 2022. Retrieved 14 January 2022.
- ↑ 2.00 2.01 2.02 2.03 2.04 2.05 2.06 2.07 2.08 2.09 2.10 2.11 2.12 2.13 2.14 2.15 2.16 2.17 2.18 2.19 2.20 2.21 2.22 2.23 2.24 2.25 2.26 2.27 2.28 2.29 2.30 2.31 2.32 2.33 2.34 2.35 2.36 2.37 2.38 2.39 2.40 2.41 2.42 2.43 2.44 2.45 2.46 2.47 2.48 2.49 2.50 2.51 "Erleada- apalutamide tablet, film coated". DailyMed. 27 October 2020. Archived from the original on 9 November 2020. Retrieved 8 November 2020.
- ↑ 3.0 3.1 "Apalutamide (Erleada) Use During Pregnancy". Drugs.com. 20 July 2020. Archived from the original on 29 November 2020. Retrieved 28 September 2020.
- ↑ 4.0 4.1 4.2 4.3 4.4 4.5 "Erleada". Archived from the original on 28 December 2020. Retrieved 14 January 2022.
- ↑ 5.0 5.1 Rathkopf DE, Morris MJ, Fox JJ, Danila DC, Slovin SF, Hager JH, et al. (October 2013). "Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer". Journal of Clinical Oncology. 31 (28): 3525–30. doi:10.1200/JCO.2013.50.1684. PMC 3782148. PMID 24002508.
- ↑ 6.0 6.1 "Espacenet - Original document". Archived from the original on 4 November 2021. Retrieved 25 August 2021.
- ↑ BNF 81: March-September 2021. BMJ Group and the Pharmaceutical Press. 2021. p. 990. ISBN 978-0857114105.
- ↑ "Erleada Prices, Coupons & Patient Assistance Programs". Drugs.com. Archived from the original on 15 January 2021. Retrieved 14 January 2022.
- ↑ 9.0 9.1 9.2 9.3 9.4 Rathkopf D, Scher HI (2013). "Androgen receptor antagonists in castration-resistant prostate cancer". Cancer Journal. 19 (1): 43–9. doi:10.1097/PPO.0b013e318282635a. PMC 3788593. PMID 23337756.
- ↑ 10.0 10.1 Kim W, Ryan CJ (February 2015). "Quo vadis: advanced prostate cancer-clinical care and clinical research in the era of multiple androgen receptor-directed therapies". Cancer. 121 (3): 361–71. doi:10.1002/cncr.28929. PMID 25236176. S2CID 6309403.
- ↑ 11.0 11.1 11.2 Kawahara, Takashi; Miyamoto, Hiroshi (2014). "Androgen Receptor Antagonists in the Treatment of Prostate Cancer". Clinical Immunology, Endocrine & Metabolic Drugs. 1 (1): 11–19. doi:10.2174/22127070114019990002. ISSN 2212-7070.
- ↑ Dellis AE, Papatsoris AG (June 2018). "Apalutamide: The established and emerging roles in the treatment of advanced prostate cancer". Expert Opin Investig Drugs. 27 (6): 553–559. doi:10.1080/13543784.2018.1484107. PMID 29856649. S2CID 46925616.
- ↑ 13.0 13.1 13.2 Patel JC, Maughan BL, Agarwal AM, Batten JA, Zhang TY, Agarwal N (2013). "Emerging molecularly targeted therapies in castration refractory prostate cancer". Prostate Cancer. 2013: 981684. doi:10.1155/2013/981684. PMC 3684034. PMID 23819055.
- ↑ 14.0 14.1 14.2 14.3 14.4 Schweizer MT, Antonarakis ES (August 2012). "Abiraterone and other novel androgen-directed strategies for the treatment of prostate cancer: a new era of hormonal therapies is born". Therapeutic Advances in Urology. 4 (4): 167–78. doi:10.1177/1756287212452196. PMC 3398601. PMID 22852027.
- ↑ Leibowitz-Amit R, Joshua AM (December 2012). "Targeting the androgen receptor in the management of castration-resistant prostate cancer: rationale, progress, and future directions". Current Oncology. 19 (Suppl 3): S22-31. doi:10.3747/co.19.1281. PMC 3553559. PMID 23355790.
- ↑ 16.0 16.1 Pinto Á (February 2014). "Beyond abiraterone: new hormonal therapies for metastatic castration-resistant prostate cancer". Cancer Biology & Therapy. 15 (2): 149–55. doi:10.4161/cbt.26724. PMC 3928129. PMID 24100689.
- ↑ 17.0 17.1 17.2 17.3 17.4 17.5 Al-Salama ZT (April 2018). "Apalutamide: First Global Approval". Drugs. 78 (6): 699–705. doi:10.1007/s40265-018-0900-z. PMID 29626324. S2CID 4653827.
- ↑ 18.0 18.1 18.2 18.3 18.4 "FDA Approves Apalutamide for Nonmetastatic Prostate Cancer". Archived from the original on 24 November 2020. Retrieved 25 August 2021.
- ↑ Anderson J (March 2003). "The role of antiandrogen monotherapy in the treatment of prostate cancer". BJU Int. 91 (5): 455–61. doi:10.1046/j.1464-410x.2003.04026.x. PMID 12603397. S2CID 8639102.
- ↑ Jeong, Se Hyo; Kim, Hun Hwan; Park, Min Young; Bhosale, Pritam Bhagwan; Abusaliya, Abuyaseer; Won, Chung Kil; Park, Kwang Il; Kim, Eunhye; Heo, Jeong Doo; Kim, Hyun Wook; Ahn, Meejung; Seong, Je Kyung; Kim, Gon Sup (January 2023). "Flavones: The Apoptosis in Prostate Cancer of Three Flavones Selected as Therapeutic Candidate Models". International Journal of Molecular Sciences. 24 (11): 9240. doi:10.3390/ijms24119240. ISSN 1422-0067.
- ↑ 21.0 21.1 21.2 21.3 21.4 21.5 Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, et al. (March 2012). "ARN-509: a novel antiandrogen for prostate cancer treatment". Cancer Research. 72 (6): 1494–503. doi:10.1158/0008-5472.CAN-11-3948. PMC 3306502. PMID 22266222.
- ↑ Ya-Xiong Tao (11 June 2014). Pharmacology and Therapeutics of Constitutively Active Receptors. Elsevier Science. pp. 351–. ISBN 978-0-12-417206-7.
ARN-509 is related structurally to enzalutamide with greater in vivo activity in CRPC xenograft models (Clegg et al., 2012).
- ↑ Joseph JD, Lu N, Qian J, Sensintaffar J, Shao G, Brigham D, Moon M, Maneval EC, Chen I, Darimont B, Hager JH (September 2013). "A clinically relevant androgen receptor mutation confers resistance to second-generation antiandrogens enzalutamide and ARN-509". Cancer Discovery. 3 (9): 1020–9. doi:10.1158/2159-8290.CD-13-0226. PMID 23779130.
- ↑ Nelson WG, Yegnasubramanian S (September 2013). "Resistance emerges to second-generation antiandrogens in prostate cancer". Cancer Discovery. 3 (9): 971–4. doi:10.1158/2159-8290.CD-13-0405. PMC 3800038. PMID 24019330.
- ↑ Moilanen AM, Riikonen R, Oksala R, Ravanti L, Aho E, Wohlfahrt G, Nykänen PS, Törmäkangas OP, Palvimo JJ, Kallio PJ (July 2015). "Discovery of ODM-201, a new-generation androgen receptor inhibitor targeting resistance mechanisms to androgen signaling-directed prostate cancer therapies". Scientific Reports. 5: 12007. Bibcode:2015NatSR...512007M. doi:10.1038/srep12007. PMC 4490394. PMID 26137992.
- ↑ Fizazi K, Albiges L, Loriot Y, Massard C (2015). "ODM-201: a new-generation androgen receptor inhibitor in castration-resistant prostate cancer". Expert Review of Anticancer Therapy. 15 (9): 1007–17. doi:10.1586/14737140.2015.1081566. PMC 4673554. PMID 26313416.
- ↑ Ivachtchenko AV, Mitkin OD, Kudan EV, Rjahovsky AA, Vorobiev AA, Trifelenkov AS, et al. (2014). "Preclinical Development of ONC1-13B, Novel Antiandrogen for Prostate Cancer Treatment". Journal of Cancer. 5 (2): 133–42. doi:10.7150/jca.7773. PMC 3909768. PMID 24494031.
- ↑ Clegg NJ, Wongvipat J, Joseph JD, Tran C, Ouk S, Dilhas A, et al. (March 2012). "ARN-509: a novel antiandrogen for prostate cancer treatment". Cancer Research. 72 (6): 1494–503. doi:10.1158/0008-5472.CAN-11-3948. PMC 3306502. PMID 22266222.
- ↑ "Archive copy" (PDF). Archived (PDF) from the original on 27 August 2021. Retrieved 25 August 2021.
{{cite web}}: CS1 maint: archived copy as title (link) - ↑ Cockshott ID (2004). "Bicalutamide: clinical pharmacokinetics and metabolism". Clin Pharmacokinet. 43 (13): 855–78. doi:10.2165/00003088-200443130-00003. PMID 15509184.
- ↑ Tran C, Ouk S, Clegg NJ, Chen Y, Watson PA, Arora V, Wongvipat J, Smith-Jones PM, Yoo D, Kwon A, Wasielewska T, Welsbie D, Chen CD, Higano CS, Beer TM, Hung DT, Scher HI, Jung ME, Sawyers CL (2009). "Development of a second-generation antiandrogen for treatment of advanced prostate cancer". Science. 324 (5928): 787–90. Bibcode:2009Sci...324..787T. doi:10.1126/science.1168175. PMC 2981508. PMID 19359544.
- ↑ Liu B, Su L, Geng J, Liu J, Zhao G (2010). "Developments in nonsteroidal antiandrogens targeting the androgen receptor". ChemMedChem. 5 (10): 1651–61. doi:10.1002/cmdc.201000259. PMID 20853390. S2CID 23228778.
- ↑ 33.0 33.1 "Apalutamide - Janssen Research and Development - AdisInsight". Archived from the original on 3 January 2019. Retrieved 25 August 2021.
- ↑ "Synthesis of thiohydantoins". Archived from the original on 5 November 2021. Retrieved 25 August 2021.
- ↑ Rathkopf DE, Morris MJ, Fox JJ, Danila DC, Slovin SF, Hager JH, Rix PJ, Chow Maneval E, Chen I, Gönen M, Fleisher M, Larson SM, Sawyers CL, Scher HI (2013). "Phase I study of ARN-509, a novel antiandrogen, in the treatment of castration-resistant prostate cancer". J. Clin. Oncol. 31 (28): 3525–30. doi:10.1200/JCO.2013.50.1684. PMC 3782148. PMID 24002508.
- ↑ Smith, M. R., Liu, G., Shreeve, S. M., Matheny, S., Sosa, A., Kheoh, T. S., ... & Small, E. J. (2014). A randomized double-blind, comparative study of ARN-509 plus androgen deprivation therapy (ADT) versus ADT alone in nonmetastatic castration-resistant prostate cancer (M0-CRPC): The SPARTAN trial. 10.1200/jco.2014.32.15_suppl.tps5100
- ↑ Bossi, A.; Dearnaley, D.; McKenzie, M.; Baskin-Bey, E.; Tyler, R.; Tombal, B.; Freedland, S.J.; Roach, M.; Widmark, A.; Dicker, A.P.; Wiegel, T.; Shore, N.; Smith, M.; Yu, M.; Kheoh, T.; Thomas, S.; Sandler, H.M. (2016). "ATLAS: A phase 3 trial evaluating the efficacy of apalutamide (ARN-509) in patients with high-risk localized or locally advanced prostate cancer receiving primary radiation therapy". Annals of Oncology. 27 (suppl_6): vi263. doi:10.1093/annonc/mdw372.52. ISSN 0923-7534.
- ↑ Chi, K.N.; Chowdhury, S.; Radziszewski, P.; Lebret, T.; Ozguroglu, M.; Sternberg, C.; Sims, R.B.; Yu, M.; Naini, V.; Darif, M.; Merseburger, A.S. (2016). "TITAN: A randomized, double-blind, placebo-controlled, phase 3 trial of apalutamide (ARN-509) plus androgen deprivation therapy (ADT) in metastatic hormone-sensitive prostate cancer (mHSPC)". Annals of Oncology. 27 (suppl_6): vi265. doi:10.1093/annonc/mdw372.54. ISSN 0923-7534.
- ↑ "Janssen Submits New Drug Application to U.S. FDA for Apalutamide (ARN-509) to Treat Men with Non-Metastatic Castration-Resistant Prostate Cancer". Archived from the original on 15 February 2018. Retrieved 25 August 2021.
- ↑ 40.0 40.1 40.2 "FDA approves new treatment for a certain type of prostate cancer using novel clinical trial endpoint". 24 March 2020. Archived from the original on 23 April 2019. Retrieved 25 August 2021.
- ↑ 41.0 41.1 41.2 41.3 "Erleada (Apalutamide): Side Effects, Dosage & Uses".
- ↑ "Archive copy" (PDF). Archived (PDF) from the original on 4 November 2021. Retrieved 25 August 2021.
{{cite web}}: CS1 maint: archived copy as title (link) - ATC code:
- CAS Number: 956104-40-8
1361232-32-7 - PubChem CID: 24872560
- DrugBank:
- ChemSpider:
- UNII:
- KEGG:
- ChEMBL:
Contents
Apalutamide
 From Mdwiki - Reading time: 18 min
From Mdwiki - Reading time: 18 min
 | |
 | |
| Names | |
|---|---|
| Trade names | Erleada, others |
| Other names | ARN-509; JNJ-56021927; JNJ-927; A52 |
| Clinical data | |
| Drug class | Nonsteroidal antiandrogen[1] |
| Main uses | Prostate cancer[2] |
| Side effects | Tiredness, joint pain, rash, falls, high blood pressure, hot flashes, diarrhea, bone fractures[2] |
| Pregnancy category | |
| Routes of use | By mouth[2] |
| External links | |
| AHFS/Drugs.com | Monograph |
| MedlinePlus | a618018 |
| Legal | |
| License data | |
| Legal status | |
| Pharmacokinetics | |
| Bioavailability | 100%[2] |
| Protein binding | Apalutamide: 96%[2] NDMA: 95%[2] |
| Metabolism | Liver (CYP2C8, CYP3A4)[2] |
| Metabolites | • NDMA[2] |
| Elimination half-life | Apalutamide: 3–4 days (at steady-state)[5][2] |
| Excretion | Urine: 65%[2] Feces: 24%[2] |
| Chemical and physical data | |
| Formula | C21H15F4N5O2S |
| Molar mass | 477.44 g·mol−1 |
| 3D model (JSmol) | |
Apalutamide, sold under the brand name Erleada among others, is a medication used to treat prostate cancer.[2] Specifically it is used in metastatic castration-sensitive prostate cancer and non-metastatic castration-resistant prostate cancer (NM-CRPC).[2] It is taken by mouth.[2]
Common side effects include tiredness, joint pain, rash, falls, high blood pressure, hot flashes, diarrhea, and bone fractures.[2] Other side effects may include stroke and seizures.[2] Use by males whose partners are pregnant may harm the baby.[2] It is a nonsteroidal antiandrogen (NSAA) and works by blocking androgens including testosterone.[4][1] As prostate cancer require androgens, blocking them slows its growth.[4]
Apalutamide was described in 2007.[6] It approved for medical use in the United States in 2018 and Europe in 2019.[2][4] In the United Kingdom 4 weeks costs the NHS about £2,700 as of 2021.[7] This amount in the United States is about 12,500 USD.[8]
Medical uses[edit | edit source]
Apalutamide is used in conjunction with castration, either via bilateral orchiectomy or gonadotropin-releasing hormone analogue (GnRH analogue) therapy, as a method of androgen deprivation therapy in the treatment of NM-CRPC.[2][9][10][11] It is also a promising potential treatment for metastatic castration-resistant prostate cancer (mCRPC), which the NSAA enzalutamide and the androgen synthesis inhibitor abiraterone acetate are used to treat.[12]
Available forms[edit | edit source]
Apalutamide is provided in the form of 60 mg oral tablets.[2] It is taken at a dosage of 240 mg once per day (four tablets) when used in the treatment of NM-CRPC.[2]
Contraindications[edit | edit source]
Contraindications of apalutamide include pregnancy and a history of or susceptibility to seizures.[2]
Side effects[edit | edit source]
Apalutamide has been found to be well tolerated in clinical trials,[13][9] with the most common side effects reported when added to surgical or medical castration including fatigue, nausea, abdominal pain, and diarrhea.[14][15][16] Other side effects have included rash, falls and bone fractures, and hypothyroidism, as well as seizures (in 0.2%), among others.[2][17][18] Apalutamide is an expected teratogen and has a theoretical risk of birth defects in male infants if taken by women during pregnancy.[2] It may impair male fertility.[2] When used as a monotherapy (i.e., without surgical or medical castration) in men, NSAAs are known to produce additional, estrogenic side effects like breast tenderness, gynecomastia, and feminization in general by increasing estradiol levels.[19] Similarly to the related second-generation NSAA enzalutamide but unlike first-generation NSAAs like flutamide and bicalutamide, elevated liver enzymes and hepatotoxicity have not been reported with apalutamide.[2]
Overdose[edit | edit source]
There is no known antidote for overdose of apalutamide.[2] General supportive measures should be undertaken until clinical toxicity, if any, diminishes or resolves.[2]
Interactions[edit | edit source]
Apalutamide has a high potential for drug interactions.[2] In terms of effects of apalutamide on other drugs, the exposure of substrates of CYP3A4, CYP2C19, CYP2C9, UDP-glucuronosyltransferase, P-glycoprotein, ABCG2, or OATP1B1 may be reduced to varying extents.[2] In terms of effects of other drugs on apalutamide, strong CYP2C8 or CYP3A4 inhibitors may increase levels of apalutamide or its major active metabolite N-desmethylapalutamide, while mild to moderate CYP2C8 or CYP3A4 inhibitors are not expected to affect their exposure.[2]
Pharmacology[edit | edit source]
Pharmacodynamics[edit | edit source]

Antiandrogenic activity[edit | edit source]
Apalutamide acts as a selective competitive silent antagonist of the androgen receptor (AR), via the ligand-binding domain, and hence is an antiandrogen.[17][21][14][9] It is similar both structurally and pharmacologically to the second-generation NSAA enzalutamide,[13][22] but shows some advantages, including higher antiandrogenic activity as well as several-fold reduced central nervous system distribution.[21][14][9] The latter difference may reduce its comparative risk of seizures and other central side effects.[21][14][9] Apalutamide has 5- to 10-fold greater affinity for the AR than bicalutamide, a first-generation NSAA.[11][10]
The acquired F876L mutation of the AR identified in advanced prostate cancer cells has been found to confer resistance to both enzalutamide and apalutamide.[23][24] A newer NSAA, darolutamide, is not affected by this mutation, nor has it been found to be affected by any other tested/well-known AR mutations.[25] Apalutamide may be effective in a subset of prostate cancer patients with acquired resistance to abiraterone acetate.[13]
Other activities[edit | edit source]
Apalutamide shows potent induction potential of cytochrome P450 enzymes similarly to enzalutamide.[2][26][27] It is a strong inducer of CYP3A4 and CYP2C19 and a weak inducer of CYP2C9, as well as an inducer of UDP-glucuronosyltransferase.[2] In addition, apalutamide is an inducer of P-glycoprotein, ABCG2, and OATP1B1.[2]
Apalutamide binds weakly to and inhibits the GABAA receptor in vitro similarly to enzalutamide (IC50 = 3.0 and 2.7 μM, respectively),[28] but due to its relatively lower central concentrations, may have a lower risk of seizures in comparison.[21][14][16]
Apalutamide has been found to significantly and concentration-dependently increase QT interval.[2]
Pharmacokinetics[edit | edit source]
The mean absolute oral bioavailability of apalutamide is 100%.[2] Mean peak levels of apalutamide occur 2 hours following administration, with a range of 1 to 5 hours.[2] Food delays the median time to peak levels of apalutamide by approximately 2 hours, with no significant changes in the peak levels themselves or in area-under-curve levels.[2] Steady-state levels of apalutamide are achieved following 4 weeks of administration, with an approximate 5-fold accumulation.[2] Peak concentrations for 160 mg/day apalutamide at steady-state are 6.0 μg/mL (12.5 μmol/L),[2] relative to peak levels of 16.6 μg/mL (35.7 μmol/L) for 160 mg/day enzalutamide and mean (R)-bicalutamide levels of 21.6 μg/mL (50.2 μmol/L) for 150 mg/day bicalutamide.[29][30] The mean volume of distribution of apalutamide at steady-state is approximately 276 L.[2] The plasma protein binding of apalutamide is 96%, while that of its major metabolite N-desmethylapalutamide is 95%, both irrespective of concentration.[2]
Apalutamide is metabolized in the liver by CYP2C8 and CYP3A4.[2] A major active metabolite, N-desmethylapalutamide, is formed by these enzymes, with similar contribution of each of these enzymes to its formation at steady-state.[2] Following a single oral dose of 200 mg apalutamide, apalutamide represented 45% and N-desmethylapalutamide 44% of total area-under-curve levels.[2] The mean elimination half-life of apalutamide at steady-state is 3 to 4 days.[2][5] Fluctuations in apalutamide exposure are low and levels are stable throughout the day, with mean peak-to-trough ratios of 1.63 for apalutamide and 1.27–1.3 for N-desmethylapalutamide.[2] After a single dose of apalutamide, its clearance rate (CL/F) was 1.3 L/h, while its clearance rate increased to 2.0 L/h at steady-state.[17] This change is considered to be likely due to CYP3A4 auto-induction.[17] Approximately 65% of apalutamide is excreted in urine (1.2% as unchanged apalutamide and 2.7% as N-desmethylapalutamide) while 24% is excreted in feces (1.5% as unchanged apalutmaide and 2% as N-desmethylapalutamide).[2]
Chemistry[edit | edit source]
Apalutamide is a structural analogue of enzalutamide and RD-162.[11][31] It is a pyridyl variant of RD-162. Enzalutamide and RD-162 were derived from the nonsteroidal androgen RU-59063, which itself was derived from the first-generation NSAA nilutamide and by extension from flutamide.[32]





History[edit | edit source]
Apalutamide was originated by the University of California system and was developed primarily by Janssen Research & Development, a division of Johnson & Johnson.[33] It was first described in the literature in a United States patent application that was published in November 2007 and in another that was submitted in July 2010.[6][34] A March 2012 publication described the discovery and development of apalutamide.[21] A phase I clinical trial of apalutamide was completed by March 2012, and the results of this study were published in 2013.[21][35] Information on phase III clinical studies, including ATLAS, SPARTAN, and TITAN, was published between 2014 and 2016.[36][37][38] Positive results for phase III trials were first described in 2017, and Janssen submitted a New Drug Application for apalutamide to the United States Food and Drug Administration on 11 October 2017.[39] Apalutamide was approved by the Food and Drug Administration in the United States, under the brand name Erleada, for the treatment of NM-CRPC on 14 February 2018.[40][18] It was subsequently approved in Canada, the European Union, and Australia.[41][4] It was the first medication to be approved specifically for the treatment of NM-CRPC.[2][17][18]
Society and culture[edit | edit source]
Names[edit | edit source]
Apalutamide is the generic name of the drug and its INN.[42][41] It is also known by its developmental code names ARN-509 and JNJ-56021927.[33][17]
Apalutamide is marketed under the brand names Erleada and Erlyand.[2][40][18][41]
Availability[edit | edit source]
Apalutamide is available in the United States, Canada, the European Union, and Australia.[2][40][18][41][4]
References[edit | edit source]
External links[edit | edit source]
| External sites: | |
|---|---|
| Identifiers: |
Original source: https://mdwiki.org/wiki/Apalutamide
Status: article is cached
 EncycloReader
is supported by the
EncycloReader
is supported by the