Antiarrhythmic agent
 From Wikidoc - Reading time: 20 min
From Wikidoc - Reading time: 20 min
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1]
Overview
[edit | edit source]Antiarrhythmic agents are a group of pharmaceuticals that are used to suppress fast rhythms of the heart (cardiac arrhythmias), such as atrial fibrillation, atrial flutter, ventricular tachycardia, and ventricular fibrillation.
While the use of antiarrhythmic agents to suppress atrial arrhythmias (atrial fibrillation and atrial flutter) is still in practice, it is unclear whether suppression of atrial arrhythmias will prolong life.[1][2]
In the past, it was believed that following myocardial infarction (heart attack), suppression of ventricular arrhythmias would prolong life. However large clinical trials found that suppression of these arrhythmias would paradoxically increase mortality,[3][4] which may happen due to the proarrhythmic effect these drugs may have (CAST trial).
In individuals with atrial fibrillation, antiarrhythmics are still used to suppress arrhythmias. This is often done to relieve the symptoms that may be associated with the loss of the atrial component to ventricular filling (atrial kick) that is due to atrial fibrillation or flutter.
-
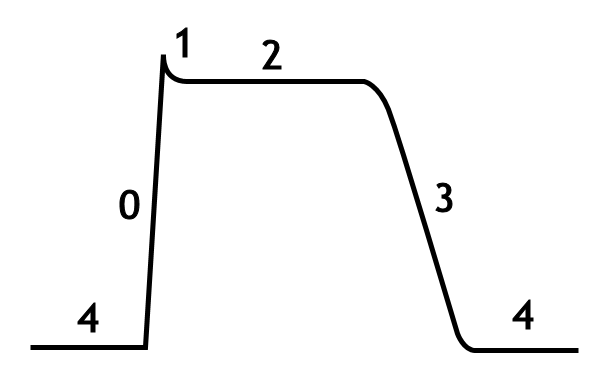 The cardiac action potential
The cardiac action potential -
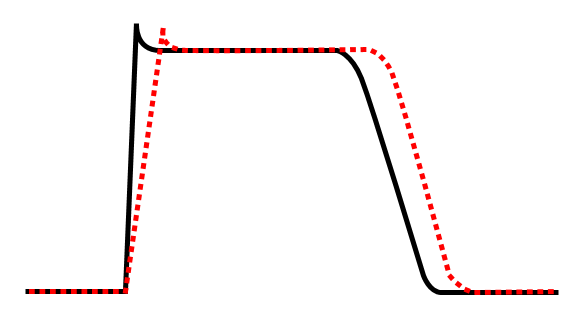 Class Ia agent decreasing Vmax, thereby increasing action potential duration.
Class Ia agent decreasing Vmax, thereby increasing action potential duration. -
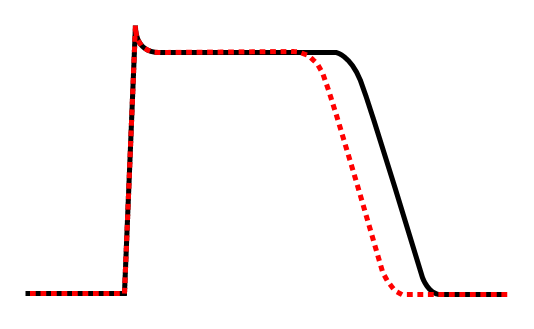 Effect of class Ib antiarrhythmic agents on the cardiac action potential.
Effect of class Ib antiarrhythmic agents on the cardiac action potential.



-
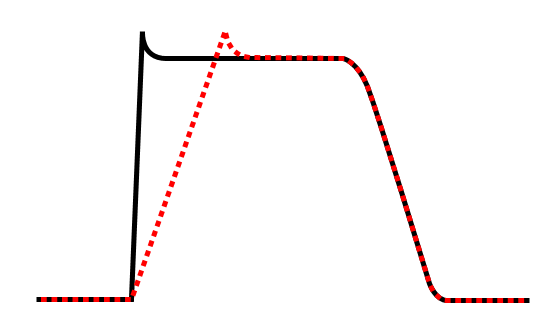 Effect of class Ic antiarrhythmic agent on cardiac action potential.
Effect of class Ic antiarrhythmic agent on cardiac action potential. -
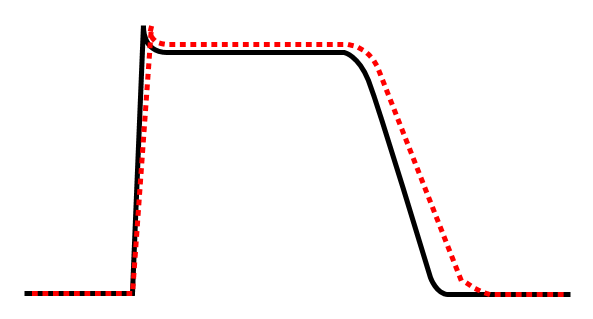 Effect of class III antiarrhythmic agent on cardiac action potential.
Effect of class III antiarrhythmic agent on cardiac action potential.


In individuals with ventricular arrhythmias, antiarrhythmic agents are often still in use to suppress arrhythmias. In this case, the patient may have frequent arrhythmic events or be at high risk for ventricular arrhythmias. Antiarrhythmic agents may be considered the first-line therapy in the prevention of sudden death in certain forms of structural heart disease, and failure of these agents to suppress arrhythmias may lead to implantation of an implantable cardioverter-defibrillator (ICD).
The use of antiarrhythmic agents in this population may be in conjunction with an ICD. In this case, the ICD is used to prevent sudden death due to ventricular fibrillation, while the antiarrhythmic agent(s) are used to suppress ventricular tachyarrhythmias so that the ICD doesn't shock the patient frequently.
Many attempts have been made to classify antiarrhythmic agents. The problem arises from the fact that many of the antiarrhythmic agents have multiple modes of action, making any classification imprecise.
Vaughan Williams Antiarrhythmic Classification
[edit | edit source]The Vaughan Williams classification, introduced in 1970,[5] is one of the most widely used classification schemes for antiarrhythmic agents. This scheme classifies a drug based on the primary mechanism of its antiarrhythmic effect. However, its dependence on primary mechanism is one of the limitations of the VW classification, since many antiarrhythmic agents have multiple action mechanisms. Amiodarone, for example, has effects consistent with all of the first four classes. Another limitation is the lack of consideration within the VW classification system for the effects of drug metabolites. Procainamide—a class Ia agent whose metabolite N-acetyl procainamide (NAPA) has a class III action—is one such example. A historical limitation was that drugs such as digoxin and adenosine – important antiarrhythmic agents – had no place at all in the VW classification system. This has since been rectified by the inclusion of class V.
There are five main classes in the Vaughan Williams classification of antiarrhythmic agents:
- Class I agents interfere with the sodium (Na+) channel.
- Class II agents are anti-sympathetic nervous system agents. All agents in this class are beta blockers.
- Class III agents affect potassium (K+) efflux.
- Class IV agents affect the AV node. They are calcium channel blockers.
- Class V agents work by other or unknown mechanisms.
Class I agents
[edit | edit source]The class I antiarrhythmic agents interfere with the sodium (Na+) channel. Class I agents are grouped by what effect they have on the Na+ channel, and what effect they have on cardiac action potentials.
- Intensity of Na+ blockade
- 1C>1A>1B (manifests as increased PR and QRS)
- Proarrhythmia may be provoked by increased heart rate. Thus common clinical practice is to perform exercise testing following drug loading.
- Pro-arrhythmic impact of Class 1C may be reversed or attenuated with the use of beta blocking adrenergic agents.
Cardiac Arrhythmia Suppression Trial (CAST)
[edit | edit source]Hypothesis: Suppression of Ventricular Premature Complexes (VPC) post-MI beneficial in terms of mortality.
Result: Flecainide and Encainide associated with unexpected increase in mortality.
Class Ia agents
[edit | edit source]Class Ia agents block the fast sodium channel. (manifests as increased PR and QRS)
Blocking this channel depresses the phase 0 depolarization (reduces Vmax), which prolongs the action potential duration by slowing conduction.
Prolongation of repolarization due to K+ channel effect. (manifests as prolonged QT interval)
Agents in this class also cause decreased conductivity and increased refractoriness.
Indications for Class Ia agents are supraventricular tachycardia, ventricular tachycardia, symptomatic ventricular premature beats, and prevention of ventricular fibrillation.
Procainamide can be used in the treatment of atrial fibrillation in the setting of Wolff-Parkinson-White syndrome, and in the treatment of wide complex hemodynamically stable tachycardias.
While procainamide and quinidine may be used in the conversion of atrial fibrillation to normal sinus rhythm, they should only be used in conjunction with an AV node blocking agent (ie: digoxin, verapamil, or a beta blocker), because procainamide and quinidine can increase the conduction through the AV node and may cause 1:1 conduction of atrial fibrillation, causing an increase in the ventricular rate.
Class Ia agents include quinidine, procainamide and disopyramide.
Class Ib agents
[edit | edit source]Class Ib antiarrhythmic agents are sodium channel blockers. Class Ib agents have fast onset and offset kinetics, meaning that they have little or no effect at slower heart rates, and more effects at faster heart rates. Class Ib agents shorten the action potential duration and reduce refractoriness. These agents will decrease Vmax in partially depolarized cells with fast response action potentials. They either do not change the action potential duration, or they may decrease the action potential duration. Class Ib drugs tend to be much more specific for voltage gated Na channels than Ia. Lidocane in particular is highly frequency dependent, in that it has more activity with increasing heart rates. This is because lidocane selectively blocks Na channels in their open and inactive states and has little binding capability in the resting state.
Class Ib agents are indicated for the treatment of ventricular tachycardia and symptomatic premature ventricular beats, and prevention of ventricular fibrillation.
Class Ib agents include lidocaine, mexiletine, tocainide, and phenytoin.
Class Ic agents
[edit | edit source]Class Ic antiarrhythmic agents markedly depress the phase 0 depolarization (decreasing Vmax). They decrease conductivity, but have a minimal effect on the action potential duration. Of the sodium channel blocking antiarrhythmic agents (the class I antiarrhythmic agents), the class Ic agents have the most potent sodium channel blocking effects.
Class Ic agents are indicated for life-threatening ventricular tachycardia or ventricular fibrillation, and for the treatment of refractory supraventricular tachycardia (ie: atrial fibrillation). These agents are potentially pro-arrhythmic, especially in settings of structural heart disease (e.g. post-myocardial infarction), and are contraindicated in such settings.
Class Ic agents include encainide, flecainide, moricizine, and propafenone.
Class II agents
[edit | edit source]Class II agents are conventional beta blockers. They act by selectively blocking the effects of catecholamines at the β1-adrenergic receptors, thereby decreasing sympathetic activity on the heart. These agents are particularly useful in the treatment of supraventricular tachycardias. They decrease conduction through the AV node.
Class II agents include esmolol, propranolol, and metoprolol.
Class III agents
[edit | edit source]Class III agents predominantly block the potassium channels, thereby prolonging repolarization.[6] Since these agents do not affect the sodium channel, conduction velocity is not decreased. The prolongation of the action potential duration and refractory period, combined with the maintenance of normal conduction velocity, prevent re-entrant arrhythmias. (The re-entrant rhythm is less likely to interact with tissue that has become refractory).
All clinically available class III drugs block IKr, not IKs
Class III antiarrhythmic agents exhibit reverse use dependent prolongation of the action potential duration (Reverse use-dependence). This means that the refractoriness of the ventricular myocyte increases at lower heart rates. This increases the susceptibility of the myocardium to early after-depolarizations (EADs) at low heart rates. Antiarrhythmic agents that exhibit reverse use-dependence are more efficacious at preventing a tachyarrhythmia than converting someone into normal sinus rhythm. Because of the reverse use-dependence of class III agents, at low heart rates class III antiarrhythmic agents may paradoxically be more arrhythmogenic.
Amiodarone is indicated for the treatment of refractory VT or VF, particularly in the setting of acute ischemia. Amiodarone is also safe to use in individuals with cardiomyopathy and atrial fibrillation, to maintain normal sinus rhythm. Amiodarone prolongation of the action potential is uniform over a wide range of heart rates so this drug does not have reverse use-dependent action.7 In contrast, dofetilide blocks only the rapid K channels; this means that at higher heart rates, when there is increased involvement of the slow K channels, dofetilide has less of an action potential-prolonging effect. 7
Sotalol is indicated for the treatment of atrial or ventricular tachyarrhythmias, and AV re-entrant arrhythmias. Ibutilide is the only antiarrhythmic agent currently approved by the Food and Drug Administration for acute conversion of atrial fibrillation to sinus rhythm.
Class III agents include amiodarone, azimilide, bretylium, clofilium, dofetilide, tedisamil, ibutilide, sematilide, and sotalol.
Class IV agents
[edit | edit source]Class IV agents are slow calcium channel blockers. They decrease conduction through the AV node, and shorten phase two (the plateau) of the cardiac action potential. They thus reduce the contractility of the heart, so may be inappropriate in heart failure. However, in contrast to beta blockers, they allow the body to retain adrenergic control of heart rate and contractility.
Class IV agents include verapamil and diltiazem.
Class V agents
[edit | edit source]Class V agents include digoxin and adenosine. Digoxin increases vagal activity via its central action on the central nervous system, thus decreasing the conduction of electrical impulses through the AV node
Major Routes of Elimination of Antiarrhythmic drugs
[edit | edit source]Hepatic: Amiodarone, Propafenone, Lidocaine, Mexilitine, Verapamil, Diltiazem.
Renal: Sotalol, Digoxin, Dofetilide, Bretylium
Mixed: Procainamide, Flecainide (70% hepatic), Quinidine (50-70% hepatic), Ibutilide, Azimilide (not FDA approved)
General Principles of Antiarrhythmic drugs
[edit | edit source]- All anti-arrhythmic drugs should be considered pro-arrhythmic.
- Pro-arrhythmia increased in presence of structural heart disease, (especially myocardial ischemia and left ventricular systolic dysfunction).
- Beta-adrenoreceptor blockers are the only drug class shown definitively to improve mortality.
- Amiodarone and Dofetilide have neutral impact on mortality following MI and in the presence of structural heart disease.
- Consider altered drug clearance in both renal and hepatic impairment and drug interactions.
Specific Drugs
[edit | edit source]Quinidine Class 1A
[edit | edit source]- Less commonly used now.
- Used in atrial and ventricular arrhythmias.
- Proarrhythmia is dose-independent and may occur with normal or even sub-therapeutic drug levels.
- Theraupetic effect determined with reference to the corrected QT interval.
- QT prolongation > 500msec should prompt abandonment of this drug class or decrease in the drug dose. Some degree of QT prolongation suggests anti-arrhythmic drug effect due to prolonged repolarization.
- Alpha-adrenergic blocking property of quinidine, in addition to peripheral vasodilation, may cause orthostatic hypotension and reflex tachycardia.
- Quinidine does not cauSe clinically important negative inotropic even in the setting of severe left ventricular dysfunction.
- The vagolytic effect of quinidine may enhance AV nodal blocking agent prior to starting quinidine.
- Avoid use of quinidine for atrial fibrillation since associated with increased mortality.
- Drug interactions
- Quinidine inhibits cytochrome P450-D6 and thus decreases the metabolism of propranolol, metoprolol, and propafenone.
- Digoxin: increased levels (may be doubled in many cases) due to decreased tissue binding (lower volume of distribution) and decreased renal and biliary clearance. Therefore Digoxin should be halved when used with Quinidine
- Warfarin: the anticoagulant effect of warfarin may be increased.
- Heparin: displaces quinidine and increases the unbound fraction of the plasma.
- Verapamil: may increase quinidine levels by decreasing metabolism.
- Amiodarone: increases quinidine levels and also has an increased impact on QT prolongation.
- Also interacts with calcium channel blockers, cimetidine, and enzyme inducing drugs (phenobarbital, phenytoin, and rifampicin).
- Side effects
- GI side effects (one third), most commonly abdominal cramping and diarrhea.
- Rash common.
- Cinchonism (hearing decrease, tinnitus, and blurred vision - may include delirium if severe).
- Thrombocytopenia and Coombe's positive hemolytic anemia. Former usually resolves within days after drug is discontinued but can persist for more than one month.
- Lupus syndrome with anti-histone antibodies.
- Granulomatous hepatitis.
- Toxic levels of quinidine cause severe QRS widening and ventricular arrhythmias. (may reverse with the infusion of sodium lactate or sodium bicarbonate).
- Proarrhythmia
- Quinidine syncope most frequently due to VT (incidence 0.5 to 4.4%).
- VF (median occurence 3 days after initiation of drug) is common to all Class 1A drugs (concordance rate ~ 30%). In other words, if Torsade de Pointes or VF is observed with one class 1A agent all other class 1A agents should be avoided due to high incidence of Torsade de Pointes.
- Because of risk of proarrhythmia all class 1A drugs should be initiated in hospital.
- If QTC > 500 msec stop drug.
Disopyramide Class 1A
[edit | edit source]- Used in atrial and ventricular arrhythmias.
- Marked negative inotropic effect related directly to plasma levels. Problematic in patients with underlying left ventricular dysfunction. Useful in patients with diastolic dysfunction or in patients with hypertrophic obstructive cardiomyopathy.
- Drug Interactions
- Phenobarbital, phenotonin, and rifamycin increase hepatic metabolism of disopyramide and decrease plasma levels.
- The negative inotropic effect of some drugs such as beta adrenoreceptor blockers is additive when used in combination with disopyramide.
- Erythromycin may increase disopyramide levels by inhibiting hepatic monoendealkalation and this has led to torsades de pointes in some patients.
- Side effects
- Anticholinergic (30%) including dry mouth, blurred vision, constipation, and urinary retention. Bladder effects typically seen in older men and can be counteracted with Bethanecol. Pyridostigmine (90 mg twelve hourly to 180 mg every eight hours) prevents or diminishes the anticholinergic effect of disopyramide and allows high tolerated doses of the drug.
- Disopyramide-induced hypoglycemia has been reported and may be due to enhanced secretion of insulin. Predisposing factors appear to include advanced age, malnutrition, and chronic renal failure. Other reported side effects includes nausea, vomiting, rash, cholestatic jaundice, and Agranulocytosis.
- Disopyramide caused regular uterine contractions in all ten near term pregnant women who were studied.
- Proarrhythmia
- As with other drugs that prolong repolarization, disopyramide may cause VF, Torsade de Pointes; although it appears to do less so than quinidine.
Procainamide Class 1A
[edit | edit source]- Useful for atrial fibrillation, atrial flutter, ventricular tachycardia, ventricular fibrillation.
- 50-60% renal excreted.
- N-acetyl-procainamide (metabolite) renal excretion (therefore adjust dose in renal impairment).
- IV dose 15-20 mg/kg.
- For each 1mg/kg dose given, blood level increases 0.5 mcg/ml.
- Acute IV use largely supplanted by amiodarone.
- Adverse effects
- 25% develop gastrointestinal side effects.
- 80% +ANA.
- 30% develop a lupus like syndrome.
- Do not check ANA unless clinically indicated.
- Pro-arrhythmia (Torsade de Pointes)
- Dose independent (similar to Quinidine).
- Infra-hisian block.
- Care with bundle branch block and intra-ventricular conduction delay.
- Accelerated ventricular response to atrial flutter (use in conjunction with AV nodal blocking agent).
Lidocaine Class 1B
[edit | edit source]- Voltage dependent sodium channel block (enhanced action in depolarized i.e. ischemic tissue).
- Therefore, useful against arrhythmias in myocardial ischemia.
- Hepatic metabolism proportional to hepatic blood flow.
- Conditions that decrease hepatic blood flow decrease clearance and increase blood levels (toxicity common with Congestive Heart Failure).
- Drug Interactions
- Propanolol and Metoprolol decrease hepatic blood flow and consequently the metabolism of lidocaine increasing plasma level by up to 80%.
- Phenobarbital decreases the plasma concentration of lidocaine.
- Side effects
- CNS effects predominate and include peri-oral numbness, paraesthesia, diplopia, hyperacusis, slurred speech, altered consciousness, seizures, respiratory arrest, and coma.
- Infra-His conduction block has been reported in some patients with pre-existing conduction system disease.
- Sinus node depression has occurred in patients with underlying sinus node disease.
- Proarrhythmia
- Clinically relevant proarrhythmias due to lidocaine are infrequent.
Mexilitine Class 1B
[edit | edit source]- Used in ventricular arrhythmias.
- Minimal hemodynamic effect including little effect on heart rate, blood pressure, cardiac output or intracardiac pressures. In patients with severe left ventricular dysfunction, mexiletine may cause increase in pulmonary capillary wedge pressure but this effect is variable.
- Ejection fraction is generally unaltered by mexiletine even in patients with structural heart disease.
- Drug interactions
- Phenytoin, phenobarbital and Rifampicin increase metabolism of Mexilitine.
- Digoxin and Warfarin not affected
- Side Effects
- GI and CNS effects most prominent which are dose and concentration related.
- Tremor usually the first sign of CNS toxicity but blurred vision, dysarthria, ataxia, and confusion may occur. Tremor may respond to beta adrenergic blocking agents.
- Thrombocytopenia reported rarely.
- Increased liver function tests and other blood dyscrasias (rare).
- Proarrhythmia: Incidence of serious proarrhythmia due to mexiletine is low (<2%).
Phenytoin Class 1B
[edit | edit source]- Used in ventricular arrhythmias.
- Hypertension may occur during intravenous administration but probably due to dilutant in the intravenous preparation.
- No significant ventricular depressant effect.
- Worsening of sinus node function especially in patients with underlying sinus disease.
- Drug Interactions
- Phenytonin induces hepatic enzymes thereby increases metabolism of numerous drugs including Quinidine, disopyramide, lidocaine, mexiletine, and Theophylline.
- Phenytoin levels are increased by Isoniazid, Chloramphenicol, disulfiram, and some sulphanamides.
- Antacids decrease absorption of Phenytoin.
- Side Effects
- CNS side effects, particularly nystagmus, and ataxia occur in a concentration dependent manner.
- Rash, nausea, blood dyscrasias, lupus syndrome, peripheral neuropathy, hyperglycemia, lymphadenopathy, Stevens-Johnson syndrome, hirsutism, and osteomalacia may also be seen.
- Gingival hyperplasia common (incidence ~ 50%). Elimination of pre-existing periodontal disease and thorough oral hygiene may prevent.
- Extravasation during IV administration may cause serious tissue damage or limb loss.
- A rare but important reaction is Phenytoin Hypersensitivity Syndrome characterized by fever, skin lesions(ranging from acne form to erythema multiforma), lymphadenopathy, hepatosplenomegaly, andleukocytosis with eosinophilia.
Flecainide Class 1C
[edit | edit source]- Used in Atrial and ventricular arrhythmias
- Heart rate typically unaffected except in the presence of sinus node disease.
- Negative inotropic effect similar to disopyramide so may exacerbate CHF in patients with left ventricular dysfunction.
- No effect on ejection fraction if EF > 50%.
- Drug interactions
- Increases Digoxin levels by 25% (via decreased clearance).
- Amiodarone and cimetidine increase flecainide levels.
- Quinidine inhibits hepatic metabolism of flecainide increases elimination half life by about 20%.
- Side effects
- CNS including blurred vision, headache, ataxia.
- CHF in patients with underlying LV systolic dysfunction.
- Proarrhythmia
- Reduced left ventricular systolic function probably also predisposes to arrhythmogenesis.
- Exercise testing recommended before hospital dismissal after achieving steady state with flecainide.
- Beta adrenergic blockade used to treat proarrhythmia due to flecainide.
- Ventricular proarrhythmia due to flecainide is rare in the treatment of paroxysmal atrial fibrillation or paroxysmal supraventricular tachycardia.
- Few cases of serious proarrhythmia reported in patients with normal hearts who receive flecainide for supraventricular arrhythmias.
Propafenone Class 1C
[edit | edit source]- Structurally similar to propranolol (also has weak β-blocking action).
- Useful for atrial fibrillation, atrial flutter, SVT.
- Cautious use in VT.
- Hepatic transformation to 5-OH propafenone by P450 2D6 enzyme.
- 7% of United States population deficient in this enzyme.
- Increased propafenone level, increased beta blocker effect, longer half-life.
- Proarrhythmia: increased in the presence of reduced ejection fraction/ischemia.
- May manifest as increased VT.
- Tachycardia provoked proarrhythmia (use dependence)
- Exercise ECG useful screening test.
- Watch for impairment of AV conduction and QRS widening.
- Increases digoxin, warfarin, and metoprolol levels.
- Not as good as ICD for preventing sudden cardiac death. Amiodarone is a useful drug in suppressing symptomatic atrial and ventricular arrhythmias but is NO SUBSTITUTE for ICD therapy in patients deemed at risk of sudden cardiac arrest.
- Thyroid toxicity
- Inhibition of T4 de-iodination to T3 leading to accumulation of rT3.
- Hypothyroid (10%); Hyperthyroid (2-12%)
- Hypothyroidism: Treat with exogenous T4 or stop amiodarone.
- Hyperthyroidism: More difficult to treat. Usually need to stop drug and consider specific anti-thyroid agents. Surgical removal of gland if unable to control ir if need to continue drug. I¹³¹ not useful.
- Pulmonary toxicity
- Always consider pulmonary toxicity with worsening CHF despite therapy.
- Diagnosis
- PFT's - low DLCO and low ρO2
- High resolution CT
- Biopsy
- Bronchoalveolar lavage
- Other side effects include photosensitivity, discoloration of the skin, and corneal micro deposits(these rarely cause a clinical problem).
- Drug interactions
- Warfarin - increased INR.
- Digoxin - increased Digoxin
- Cyclosporine - increased Cyclosporine
- Proarrhythmia
- Amiodarone may cause life threatening ventricular arrhythmias including ventricular fibrillation and Torsade de Pointes.
- Less pro-arrhythmic than other antiarrhythmic agents.
- Clinical trials
- Canadian Amiodarone Myocardial Infarction Arrhythmia Trial (CAMIAT): This trial randomized 1,202 patients after myocardial infarction who had ten or more ventricular premature depolarizations per hour to amiodarone. Over a 1.79 year follow up, resuscitative ventricular fibrillation or arrhythmic death was less in the patients treated with amiodarone. It was concluded that amiodarone reduces ventricular fibrillation arrhythmic death after myocardial infarction with frequent or repetitive ventricular premature complexes. The absolute risk reduction was greater in patients with previous myocardial infarction or congestive heart failure.
- EMIAT: The EMIAT Trial randomized 1,486 patients with ischemic ventricular dysfunction after myocardial infarction, ejection fraction less than 40%. Over a 21 month follow up, all cause and cardiac mortality was similar in both groups. There was a 35% decrease in arrhythmic death in patients receiving amiodarone. Amiodarone therapy in this group of patients was not supported by the study. The conclusions of these trials differ as did their entry trials. CAMIAT focused on ventricular ectopy whereas EMIAT focused on ventricular dysfunction. Clearly, patients in EMIAT were sicker with substantially greater overall mortality. Interestingly, the details of CAMIAT indicate that like EMIAT all cause mortality was unaffected by amiodarone. The ultimate message form these trials is ambiguous but amiodarone at the very least was shown to be no worse than placebo supporting at minimum its use in patients who require antiarrhythmic therapy for other reasons.
- JESSICA Trial.
- CHF Trial.
- RECOMMENDATIONS FOR ROUTINE LABORATORY TESTING WITH CHRONIC AMIODARONE THERAPY
- Thyroid Function Tests (TSH and T4) - Baseline, 3 months and every 6 months
- Liver Function Tests - Baseline and every 6 months
- Chest X-ray - Baseline and yearly
- Pulmonary Function Tests - Baseline and if symptoms develop or change
- Ophthalmic evaluation - Baseline if visual symptoms or for symptom changes
- Pure IKr blocker
- Approved for atrial fibrillation and atrial flutter
- Inpatient loading mandatory due to risk of Torsades de Pointes VT.
- Dosing adjusted by QT interval change (at rest).
- Careful dosing in renal insufficiency (80% renally excreted).
- 20% liver (cytochrome P450)
- Initiation and Dosing
- Plan on atleast 3 days hospitalization or 12 hours after conversion to normal sinus rhythm. (whichever is greater)
- Withdraw/avoid: verapamil, cimetidine, trimethoprim, ketoconazole, prochlorperazine.
- Get potassium >4.0
- Measure QTc: if >440 msec (500 msec w/Intraventricular conduction delay) find another drug.
- Blocks IKr and slow inactivation of sodium channels (increases atrial refractoriness).
- Mostly used for acute termination of atrial fibrillation and to facilitate cardioversion in resistant cases.
- Good for WPW.
- Risk of Torsades de Pointes VT is 2-3%; higher with advanced LV dysfunction (EF<20%).
- Adjunctive use of magnesium after ibutilide may prevent proarrhythmia.
- Chemically similar to Amiodarone but has no Iodine (lower risk of pulmonary, liver and thyroid complications).
- Shorter elimination half-life (24 hours versus weeks).
- Indicated for atrial Flutter and atrial fibrillation (not ventricular arrhythmias or sudden death prevention).
- More effective than placebo in maintaining sinus rhythm.
- The efficacy of dronedarone in patients with atrial fibrillation is superior to placebo but appears inferior to amiodarone and sotalol.
- Reduces hospitalization due to cardiovascular events or death.
- Reduces rate of strokes.
- Contraindicated in
- NYHA class IV heart failure
- NYHA II-III heart failure with recent decompensation requiring hospitalization.
- In the ANDROMEDA study dronedarone doubled the death rate in patients with moderate to severe heart failure although the precise mechanism is unknown. Because of these findings, dronedarone is contraindicated in class IV CHF or in patients with recent exacerbation of heart failure.
- Proarrhythmia
- The pro-arrhythmic potential of dronedarone is low and similar to amiodarone.
- Common side effects include bradycardia and QT interval prolongation (class III effect) along with nausea, rash and increased creatinine.
- Clinical trials
- EURIDIS and ADONIS trials reported that when compared with placebo dronedarone was superior in maintaining sinus rhythm after electrical, pharmacologic or spontaneous conversion to sinus rhythm from atrial fibrillation or flutter with no (short term) differences in lung and thyroid function
- ANDROMEDA study reported a significant increase in the rate of death in patients with moderate to severe congestive heart failure.
- ATHENA trial reported that dronedarone was significantly more effective than placebo in reducing the composite endpoint of first hospitalization due to cardiovascular events or death with a significant reduction in the rate of cardiovascular death, but not in the rate of death from any cause. A post-hoc analysis of the ATHENA- trial also showed a significant reduction in the rate of stroke although the clinical significance of this is unclear.
Bretylium Class III
[edit | edit source]- Used in ventricular arrhythmias (mostly VF).
- Bretylium rarely used and then only in the setting of recurrent VT or VF having failed lidocaine, procainamide, and DC cardioversion.
- Bretylium causes initial norepinephrine release with subsequent depletion of norepinephrine from sympathetic nerve terminals. This results in increased blood pressure followed by orthostatic hypotension.
- Side effects
- Orthostatic hypotension which may take several days to resolve.
- Nausea
- Vomiting during delivery of initial dose.
- Proarrhythmia: Initial worsening of arrhythmias due to release of norepinephrine.
Sotalol Class III
[edit | edit source]- Used in Atrial and ventricular arrhythmias
- Potent beta adrenergic blocker so may cause bradycardia and worsening of left ventricular dysfunction.
- Anti-hypertensive effect.
- Drug interactions
- Beta blocking action may cause exacerbate effect of drugs that have AV nodal inhibitory effects such as digoxin or non vascular selective calcium channel blockers such as verapamil and diltiazem.
- Side effects
- Most side effects related to beta blocking action
- Fatigue most prominent.
- Proarrhythmia
- QT lengthening can result in ventricular fibrillation or Torsade de pointes.
- Proarrhythmia rates of 3-5% have been reported.
- Care in patients with Hypokalemia/Hypomagnesia (i.e.those taking diuretics) and use of higher dose ranges (greater than 160 mg twice daily) are associated with greater risk of proarrhythmia.
- SWORD Trial (Survival With Oral D Sotalol) evaluated D sotalol (a pure Ikr potassium channel blocker with no beta blocking action) in 3,121 patients with LV dysfunction and recent myocardial infarction or recent heart failure and a remote myocardial infarction.
- Use of D-sotalol was associated with increased mortality over placebo.
- Still commonly used.
- Effects mediated via central and peripheral augmentation of vagal tone.
- Direct action on atria and AV node only at toxic concentrations.
- Increases sympathetic tone and Calcium loading leading to increased automaticity and delayed afterdeploarisations.
- Electrophysiology
- Inotropic action via inhibition of Na-K ATPase and Na-Ca exchange.
- Minimal effects on surface ECG at therapeutic doses.
- May improve conduction in some accessory pathways (avoid in WPW syndrome).
- No effect on rate of conversion to sinus rhythm in patients with acute episode of atrial fibrillation.
- Rarely effective as sole agent for rate control. Must confirm heart rate control by exercising (aim for heart rate less than 110 with moderate walking).
- Clinical Pharmacology
- 60-80% oral bioavailability (decreased with cholestyramine, antacids, colestipol).
- P-glycoprotein: excretes absorbed digoxin back in to gut lumen (also controls renal elimination).
- Drug interactions
- Quinidine: Increase digoxin levels by displacing from binding sites and inhibiting P-glycoprotein system.
- Amiodarone, Propafenone, Verapamil: decrease renal and non-renal clearance.
- Cyclosporine, anti-fungals, benzodiazepines: inhibit P-glycoprotein.
- Dosing guided by efficacy, adverse effects, ECG intervals and serum levels (rule of thumb: do not exceed dose that prolongs QRS more than 25% or QT >550ms)\
- Toxicity
- Non-cardiac - anorexia, nausea, vomiting, changes in color vision including scotoma, halo vision and altered color perception.
- Cardiac - due to exaggerated effects on sinus and AV node and intracellular Ca loading causing delayed afterdeploarizations.
- Arrhythmias: almost any, but high grade AV block with accelerated junctional rhythm or bi-directional VT strongly suggestive.
- Nausea (common)
- Vomiting (common)
- Anorexia (common)
- Diarrhea (common)
- Thrombocytopenia (occasionally)
- Marrow suppression (occasionally)
- Lupus (rare)
- Fever
- Pro-arrhythmia (0.5-8%) - dose independent - may occur soon after initiation (i.e. when plasma levels low)
- Activates IK Ado shortens action potential duration (direct effect).
- Decreases cAMP, decreased delayed afterdeploarizations, early afterdepolarizations (indirect effect).
- All actions mediated via A1 receptor and G proteins (not present in ventricle).
- Extensive first pass metabolism.
- Re-uptake blocked by dipyridamole.
- Methylxanthines block A1 receptor.
- Drug interactions
- Dipyridamole: increases effect.
- Methylxanthines: No effect.
- Transplanted heart: increases effect.
EKG Examples
[edit | edit source]Shown below are EKG tracings showing the effect of adenosine on AV conduction, but in addition the effect on atrial refractoriness. The first tracing is clearly atrial flutter with an atrial rate of 270/min, and a ventricular rate of 135/min. The second recording shows much faster atrial activity. The second tracing is either atrial fibrillation (a know result of adenosine) or a faster atrial flutter (type II flutter) with variable block. Both of these effects could be seen as a result of the decrease in the atrial refractory period by the stimulation of the adenosine.


Copyleft images obtained courtesy of ECGpedia, http://en.ecgpedia.org
Shown below is an EKG demonstrating a supraventricular tachycardia. It is a somewhat unusual presentation for someone with angina. The arrhythmia terminated with adenosine which has a powerful cholinergic effect that blocks conduction through the AV node.

Copyleft image obtained courtesy of ECGpedia, http://en.ecgpedia.org
IN HOSPITAL VERSUS OUTPATIENT INITIATION OF ANTI-ARRHYTHMIC DRUGS
[edit | edit source]The decision to initiate anti-arrhythmic drug therapy in hospital versus the outpatient setting is determined largely by likelihood of risk versus cost and inconvenience of inpatient drug loading. In the case of Dofetilide in-hospital initiation is mandated. Unfortunately, few prospective data are available regarding safety of outpatient initiation of AAD therapy. In the case of atrial fibrillation, outpatient loading should be avoided in patients with symptomatic sinus node dysfunction, AV conduction disturbance, bundle branch block, structural heart disease and QT prolongation. The most important risk of AAD therapy is that that of proarrhythmia, most commonly due to Torsades de Pointes, but also virtually any other arrhythmia such as ventricular tachycardia and atrial arrhythmias.
- In general, in the absence of significant bradycardia and with normal ventricular function, QRS and QT intervals, proarrhythmia risk is low.
- Unsuspected sinus node dysfunction that is suppressed by AA agent may lead to prolonged sinus node recovery with termination of AF leading to possible syncope.
- Propafenone and flecainide also worsen AV node and His Purkinji conduction thus if outpatient use is contemplated initial cardioversion in hospital is prudent.
- For Sotalol, outpatient initiation is probably safe if baseline QT interval is less than 450ms in the absence of renal dysfunction and risk factors for Torsade de Pointes.
- Amiodarone is generally considered to be safe to administer as an out-patient. Risk of pro-arrhythmia is low.
- In hospital administration of Dofetilide is mandatory.
In most cases, AAD should be started at the lowest possible dose and titrated upwards and therapeutic efficacy monitored with reference to PR interval (flecainide, propafenone, sotalol and amiodarone), QRS (flecainide, propafenone) and QT intervals (sotalol, amiodarone and disopyramide) at rest (sotalol) or with exercise (Class IC agents). Measurement of serum drug levels is rarely helpful.
Choice of agent in PSVT
[edit | edit source]- Hypotension, Uncertain diagnosis (especially if WCT), Heart failure - Adenosine
- Dipyridamole, Theophyllines, Bronchoconstriction, Transplanted heart, PSVT recurs post adenosine - Verapamil/Diltiazem
Trials
[edit | edit source]The Cardiac Arrhythmia Suppression Trial[7] was a medical study that demonstrated that some class 1 anti-arrhythmic drugs were not helpful to people suffering from ventricular premature depolarization. Specifically, for populations of recent heart attack survivors, the study observed lower mortality in a control population than in populations treated with encainide, flecainide or moricizine (all class 1c drugs).
Mnemonics
[edit | edit source]Mnemonic for Class I-IV agents: SoBe PoCa (SOBE as in South Beach or the drink, POCA as in Polka)
Sodium channel blockers, Beta blockers, Potassium channel blockers, Calcium channel blockers
Related Chapters
[edit | edit source]References
[edit | edit source]- ↑ Wyse D, Waldo A, DiMarco J, Domanski M, Rosenberg Y, Schron E, Kellen J, Greene H, Mickel M, Dalquist J, Corley S (2002). "A comparison of rate control and rhythm control in patients with atrial fibrillation". N Engl J Med. 347 (23): 1825–33. PMID 12466506.
- ↑ Nichol G, McAlister F, Pham B, Laupacis A, Shea B, Green M, Tang A, Wells G (2002). "Meta-analysis of randomised controlled trials of the effectiveness of antiarrhythmic agents at promoting sinus rhythm in patients with atrial fibrillation". Heart. 87 (6): 535–43. PMID 12010934.
- ↑ "Preliminary report: effect of encainide and flecainide on mortality in a randomized trial of arrhythmia suppression after myocardial infarction. The Cardiac Arrhythmia Suppression Trial (CAST) Investigators". N Engl J Med. 321 (6): 406–12. 1989. PMID 2473403.
- ↑ "Effect of the antiarrhythmic agent moricizine on survival after myocardial infarction. The Cardiac Arrhythmia Suppression Trial II Investigators". N Engl J Med. 327 (4): 227–33. 1992. PMID 1377359.
- ↑ Vaughan Williams EM. "Classification of anti-arrhythmic drugs." In: Symposium on Cardiac Arrhythmias, Sandfte E, Flensted-Jensen E, Olesen KH eds. Sweden, AB ASTRA, Södertälje, 1970;449-472.
- ↑ Lenz TL, Hilleman DE, Department of Cardiology, Creighton University, Omaha, Nebraska. Dofetilide, a New Class III Antiarrhythmic Agent. Pharmacotherapy 20(7):776-786, 2000. (Medline abstract)
- ↑ Entry on Clinical Trials.gov
Template:Antiarrhythmic agents Template:Major Drug Groups
 EncycloReader
is supported by the
EncycloReader
is supported by the