Categories
Cardiomyopathy pathophysiology
 From Wikidoc - Reading time: 21 min
From Wikidoc - Reading time: 21 min
|
Cardiomyopathy Microchapters |
|
Diagnosis |
|---|
|
Treatment |
|
Guidelines |
|
2020 AHA/ACC Guideline for the Diagnosis and Treatment of Patients With Hypertrophic Cardiomyopathy |
|
Case Studies |
|
Cardiomyopathy pathophysiology On the Web |
|
American Roentgen Ray Society Images of Cardiomyopathy pathophysiology |
|
Risk calculators and risk factors for Cardiomyopathy pathophysiology |
Editor-In-Chief: C. Michael Gibson, M.S., M.D. [1] ; Associate Editor(s)-in-Chief: Lina Ya'qoub, MD; Olubadewa A. Fatunde, MD, MPH; Edzel Lorraine Co, DMD, MD[2]
Overview
[edit | edit source]The different etiologies of cardiomyopathy (CM), resulting in abnormal heart structure and function are myriad. Our knowledge of this disease entity has progressed significantly since the term was first used in 1957. Historically, CM has been grouped in three different categories by phenotype or symptomatic presentation (later confirmed through echocardiographic and autopsy studies), ranging from dilated to restrictive to hypertrophic forms of CM. Emerging additional categories include arrhythmogenic right ventricular dysplasia/cardiomyopathy (ARVD/C) and unspecified CM. Dilated and Hypertrophic CM, which share symptoms of left heart failure, can be distinguished by a patient's ejection fraction (EF), left ventricular (LV) wall thickness, and LV end diastolic volume (LVEDV). Restrictive CM is relatively uncommon and presents largely with symptoms of right-sided heart failure (HF) and diastolic dysfunction. Some etiologies (e.g. inherited metabolic disorders, sarcoidosis, hemachromatosis, etc.) may cause more than one type of CM. This overlap in classification underscore the limitations of phenotypic classification system. For this reason, and with improving technology, the American Heart Association proposed a classification of CM emphasizing primary and secondary (to other systemic diseases) etiologies. Primary CM is subdivided into genetic, acquired, and mixed causes.[1] Genetic cause include HCM, ARVD/C, ion channel disorders, storage and infiltrative diseases. Genetics play an important and increasing role in the pathophysiology of CM. The genetic basis of hypertrophic cardiomyopathy (HCM) is well established. Approximately 30% of Dilated CM (DCM) cases are familial. Mutations in over 40 different genes have been described (locus heterogeneity). Various mutations within those genes have produced CM (allelic heterogeneity), and the same mutation can manifest differently within different family members (incomplete penetrance). The clinical presentation of patients with cardiomyopathy can vary widely, depending on the underlying mechanism of disease. Symptoms range from exercise intolerance and progressive heart failure to fatal arrhythmias and sudden cardiac death. Currently, treatment of CM is driven primarily by phenotype. Therefore, the discussion below will focus on this classification.
Cardiomyopathy Pathophysiology
[edit | edit source]In 2006, the American Heart Association defined cardiomyopathies as:[2]
"...a heterogeneous group of diseases of the myocardium associated with mechanical and/or electrical dysfunction that usually (but not invariably) exhibit inappropriate ventricular hypertrophy or dilatation and are due to a variety of causes that frequently are genetic. Cardiomyopathies either are confined to the heart or are part of generalized systemic disorders, which may lead to cardiovascular death or progressive heart failure-related disability." [1]
Table 1 below lists the various gene products implicated in cardiomyopathy.
| Genes impacting cardiomyopathy | Normal function/Consequence of abnormal gene product |
|---|---|
| Sarcomeric proteins |
|
| Dystrophin complex |
|
| Desmosome complexes
(Desmos - "bond", Soma - "body") |
|
Remaining cytoskeletal proteins
|
Integrate & stabilize myocyte
|
| Enzyme Deficiency | |
| Mitochondrial deficiency |
|
| Heritable systemic diseases (no mutation of heart genes) |
The most common modes of transmission is Autosomal Dominant, followed by X-linked inheritance. Most known inherited genetic defects to date are associated with hypertrophic CM. However, research is uncovering more and more genetic associations with dilated CM. Missense mutations are most common among all forms of CM; nonsense and frameshift mutations also contribute to dysfunctional structural and cellular metabolism proteins (see Table 1 above) causing CM. With the exception of dystrophinopathies, deletions are relatively rare. [1][2]

The mutations in sarcomere genes (coding for actin, myosin, titin, etc.) are the best characterized to date. A normal sarcomere is pictured above in Figure 1. Mutations in Titin, a large sarcomeric protein that maintains structure & participates in signaling, are the most common cause of dilated CM (approximately 20% of known cases). Severity of clinical disease is usually commensurate with an increasing number of mutations.[2] Genetics will continue to play an increasing role in diagnosis and management of CM.
DILATED CARDIOMYOPATHY
[edit | edit source]Dilated CM is the most common CM, comprising approximately 90% of all cardiomyopathies. [3] [4] The many causes of Dilated CM all share the following phenotype: Enlarged heart, decreased systolic function. This results from a failed attempt to ration increasingly insufficient resources following myocyte injury pictured below, in Figures 2 and 3:

Figure 3, below, expounds in detail on the failed attempts of the heart to adjust to significant myocardial injury, underlying the dilated CM phenotype.

Only one-third of all etiologies of dilated CM have been fully characterized, many of which have a genetic etiology. Two-thirds of dilated CM remain classified as idiopathic, reflecting the need for further investigation. Many postulate a yet undiscovered genetic basis for many of these CM.[1][2][3][4]
Important etiologies of Dilated CM are listed below in Table 2.
| Subtype | Example | Notes |
|---|---|---|
| Inflammatory Myocarditis | (Mechanism of injury: Direct invasion → production of cardiotoxic substances → chronic inflammation without persistent infection) | |
| Infective | Viral/Parasitic/Bacterial/Fungal/Spirochetal/Ricketsial/Fungal | |
| Non-infective | Granulomatous inflammatory diseases/Eosinophilic myocarditis/Hypersensitivity myocarditis/Polymyositis-Dermatomyositis/Collagen Vascular Disease/Pregnancy/Transplant Rejection | |
| Toxic | Alcohol | Most common etiology, comprising >10% of cases of DCM |
| Catecholamine | Amphetamines, Cocaine, Pheochromocytoma, Stress-induced CM (Takotsubo's) | |
| Chemotherapeutic agents | Adramycins, Tyrosine Kinase Inhibitors, immune checkpoint inhibitors | |
| IFN | ||
| Other therapeutic agents | Hydroxychloroquine, Chloroquine, Lithium, phenothiazine antipsychotics, antiretroviral therapies | |
| Drugs of misuse | emetine (Ipecac), anabolic steroids | |
| Heavy metals | lead; mercury (amalgam fillings, cinnabar); cobalt (tainted beer production) | |
| Occupational exposure | hydrocarbons, arsenicals | |
| Metabolic | Nutritional deficiencies | |
| Electrolyte deficiencies | ||
| Endocrinopathy | ||
| Obesity | ||
| Hemochromatosis | ||
| Inherited Metabolic Pathway Defects | Familial (30%) | Skeletal & Cardiac myopathy/Dystrophin-related dystrophy (Duchenne's, Becker's) - X-linked/Mitochondrial myopathies (e.g. Kearns-Sayre syndrome)/Arrhythmogenic ventricular dysplasia/Hemochromatosis/associated with other systemic disease/Susceptibility to immune-mediate myocarditis |
| Overlap with Nondilated Cardiomyopathy | "Minimally dilated CM"/Hemochromatosis/Amyloidosis/Hypertrophic CM | |
| Miscellaneous (Shared Elements of Above Etiologies) | Arrhythmogenic Ventricular (RV>LV) dysplasia | |
| LV Noncompaction | ||
| Peripartum | ||
| Tachycardia-related cardiomyopathy | ||
| LBBB | ||
Chronic myocarditis is the oldest known cause of cardiomyopathy, described in literature as 'heart muscle disease,' as far back as the mid-1850s.
- Inflammatory Myocarditis
- Infective etiologies - common pathway: Direct invasion, production of cardiotoxic substances, chronic inflammation without persistent infection. [2][3][4]
- Viral
- Epidemiology:
- Most common viral causes of myocarditis: Coxsackievirus, Adenovirus, HIV, hepatitis C virus
- Prior to HAART, HIV represented 1-2% of cases of dilated cardiomyopathy
- At present, HIV may interact with other viruses to produce "multiple-hits" to the myocardium and increase susceptibility to disease.
- Hep C is a major cause of myocarditis & DCM, particularly in endemic countries
- Additional implicated viruses:
- RNA viruses: enterovirus, echovirus, polioviruses. Influenza virus (winter & spring)
- DNA viruses: Herpesviruses (Varicella zoster, CMV, EBV, HHV6), Parvovirus B19
- May infect vascular endothelial cells
- Most common viral causes of myocarditis: Coxsackievirus, Adenovirus, HIV, hepatitis C virus
- Less common viral causes of myocarditis (often developing settings): Mumps, RSV, dengue & yellow fever, Lassa fever
- Symptoms:
- Most common presentation are signs and symptoms of HF
- Patients can also present with chest pain or acute MI
- More rarely, tachyarrythmias (atrial or ventricular) or thromboembolic manifestations can occcur
- Figure 4, below depicts the specific mechanism with which common viruses above lead to dilated CM.
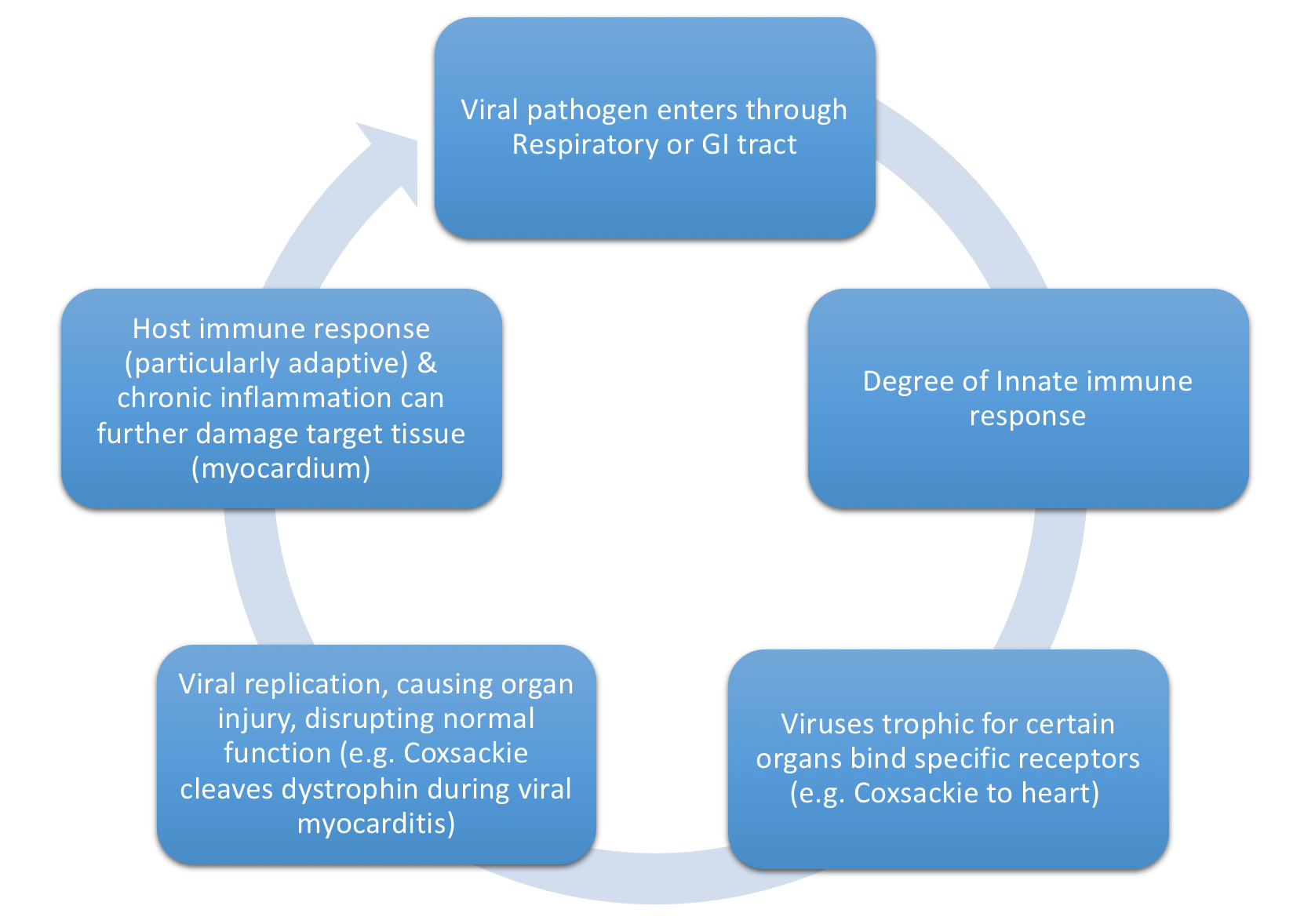
Figure 4. Mechanism through which viruses can cause dilated CM. - Activated viral proteases (e.g enteroviral protease 2A) can activate host tyrosine kinases to facilitate further viral entry as well as facilitate viral replication and infection through degradation of dystrophin
- Innate immune response depends on Toll-like receptors to recognize common antigenic patterns
- Initial immune response critical to limiting viral injury
- Early immunosuppresion can increase viral replication & worsen cardiac injury
- Timely downregulation of resultant adaptive immune response also important to prevent autoimmune injury
- Ongoing cytokine release activates matrix matalloproteinases (MMP)
- Inappropriately high levels of MMP can destroy the collagen & elastin cytoskeleton, potentially leading to a dilative CM physiology
- Initial immune response critical to limiting viral injury
- Fulminant viral myocarditis is rapid progression (days) from a severe febrile respiratory illness to cardiogenic & multiorgan shock (including renal & hepatic failure, coagulopathy).
- This often improves with appropriately aggressive supportive care.
- Epidemiology:
- Parasitic
- Chagas' disease (autonomic dysfunction, microvascular damage → CV (SA/AV node dysfcn & RBBB, Thrombogenic small ventricular aneurysm esp at apex) & GI disease. [3][4]
- Most common infective cause of cardiomyopathy
- Third most common parasitic infection in the world
- Named after the Brazilian physician, Dr. Carlos Chagas, who discovered the disease in 1909
- Caused by Trypanosoma cruzi.
- Transmitted most commonly by Reduvid bug in South & Central America > Also Organ donation, Vertical transmission (mother to baby), oral.
- Mechansim of action (MOA): Both direct & indirect mechanisms
- Direct - Parasite → myocyte lysis & neuronal damage
- Indirect - Chronic immune system activation
- 2 phases:
- Acute phase - Parisitemia, 95% of patients are asymptomatic. Others present with nonspecific, acute myocarditis and meningoencephalitis
- Silent phase - Progress slowly in over 10-30yrs, end with Heart failure & GI symptoms. 5 yr survival <30% once CHF symptoms begin.
- African Trypanosomiasis
- Also known as “sleeping sickness”
- Transmitted by Tsetse fly, only found in rural regions of African countries
- 2 forms:
- West African form due to Trypanosoma brucei gambiense (98% of reported cases)
- East African form - Trypanosoma brucei rhodensiense.
- Same two clinical phases listed above for Trypanosoma cruzi. If untreated, both will lead to coma and death.
- East African form (rhodensiense) more aggressive. Death in months without treatment. Likely under-reported for this reason.
- West African form (gambiense), can persist in acute phase last 1-2 years. Total infection may last up to 7 years untreated, but often kills by 3 years.
- MOA: Perivascular infiltration → Myocarditis & heart failure with frequent arrhythmias.
- Toxoplasmosis
- Transmitted by undercooked pork, Cat feces, organ transplant, transfusion, Vertical Transmission (~T. cruzi)
- Immunocompromised, more likely to have reactivation of latent infection from cysts
- Symptoms (Sx): Myocarditis, pericardial effusion, constrictive pericarditis, CHF + chorioretinitis + encephalitis
- Trichinellosis
- Trichinella larvae migrate into skeletal Mm → myalgias, weakness, fever
- Trichinellosis : Trichinella spiralis larva found in uncooked meat :: Toxoplasmosis : found in undercooked meat, Cat litter
- CHF occurs secondary to a eosinophilic inflammatory response
- Chagas' disease (autonomic dysfunction, microvascular damage → CV (SA/AV node dysfcn & RBBB, Thrombogenic small ventricular aneurysm esp at apex) & GI disease. [3][4]
- Bacterial (Diphtheria 1/2 of all cases of bacterial myocarditis. B-hemolytic strept, Tuberculosis, T. whippeli)
- Diphtheria releases a toxin that impairs protein synthesis & disrupts the conduction system.
- Rx c Antitoxin
- ß-hemolytic strept associated with Rheumatic Fever → inflammation & fibrosis of cardiac valves and systemic tissue.
- Diphtheria releases a toxin that impairs protein synthesis & disrupts the conduction system.
- Spirochetal (Borrelia burgdoferi -- Lyme disease)
- Conduction disease lasting 1-2 weeks after Abx
- Rickettsial (Q fever)
- Fungal (Rare. Most commonly Candida, via hematogenous spread or direct invasion.)
- Viral
- Noninfective[3][4]
- MOA: secondary to Antibodies & Cytokines from prior physical injury & viral infection
- Granulomatous inflammatory disease - Ventricular arrhythmias & conduction blocks in a patient with concurrent CHF & chest pain syndrome. Idiopathic.
- Sarcoidosis
- Patients with Pulmonary Sarcoidosis at higher risk for cardiac involvement
- Thought to have an infectious or environmental trigger, as regional clustering of cases
- Inflammation resolves into areas of fibrosis that can serve as nidus for reentrant circuits.
- Fewer granulomas associated with better prognosis.
- Giant Cell myocarditis
- Rapidly progressive HF and tachyarrhythmias
- Comprise 10 - 20% of cases of biopsy positive myocarditis
- Path on endomyocardial biopsy: extensive eosinophilic infiltration
- A/w thyomomas, thyroiditis, pernicious anemia, other AID
- Rapid deterioration, urgent transplantation
- Sarcoidosis
- Infective etiologies - common pathway: Direct invasion, production of cardiotoxic substances, chronic inflammation without persistent infection. [2][3][4]

- Eosinophilic myocarditis
- Etiologies varied, including Eosinophilic granulomatosis with polyangiitis (EGP, formerly Churg-Strauss), antecedent infection (Mediterranean & African countries), and malignancies.
- Hypersensitivity myocarditis
- Path on endomyocardial biopsy: infiltration with lymphocytes ad mononuclear cells with a high proportion of eosinophils.
- Chronic Antibiotics, most common etiology.
- Others, include Thiazides, Anticonvulsants, Indomethacin, Methyldopa.
- Polymyositis, dermatomyositis
- Skeletal & Cardiac Muscle affected
- Collagen vascular disease
- associated with Pericarditis, Vasculitis, Pulmonary hypertension, accelerated CAD
- Pregnancy
- Peripartum cardiomyopathy (PPCM)
- Definition - imprecise due to varied criteria across many international societies. 2010 European Society of Cardiology's listed below.
- 3rd trimester through 6 months postpartum, with no prior cardiac disease (often presumed)
- Absence of another identifiable cause for the HF.
- LVEF <45%. The LV may or may not be dilated.
- Incidence varies across the world. Limited by incomplete data.
- Highest in 1:100 in Zaria, Nigeria, due to the Hausa tribe's (predominantly located in Northern Nigeria) custom of eating kanwa, a dry lake salt, for forty days after delivery
- Lowest in Japan, 1:20,000 live births
- Other countries with known estimates:
- 1:300 in Haiti
- 1:1000 in South Africa
- around 1 in 1000 to 4000 live births in the United States
- 1:2400 in Canada
- 1:5719 in Sweden
- 1:10,149 in Denmark
- RF: increase maternal age, increase parity, twin pregnancy, malnutrition, Tocolytics for premature labor, Preeclampsia
- Etiology Multifactoral:
- Known association with anti-angiogenic signaling (a process exacerbated by preeclampsia)
- Mice lacking cardiac PGC-1α, a regulator of pro-angiogenic factors (e.g. VEGF), develop severe DCM
- Studies investigating proangiogenic agents (VEGF with bromocriptine) as potential therapies.
- Additionally proposed mechanisms associated with path findings of lymphocytic myocarditis on endomyocardial biopsy:
- increased susceptibility to viral myocarditis
- cross-reactivity of anti-uterine antibodies vs. cardiac muscle causing an autoimmune myocarditis
- microvascular angiogenic imbalance within myocardium
- abnormal prolactin cleavage fragment, induced by oxidative stress → Myocardial apoptosis
- Inflammatory cytokines
- TNF-alpha & interleukin-6 elevated in PPCM compared with controls
- Increased levels of C-reactive protein & Fas/Apo-1 (apoptosis signaling receptor) a/w more severe disease
- Genetic predisposition
- sarcomeric protein mutations: TTN-truncating variant (gene encoding Titin)
- Guanine nucleotide-binding proteins beta-3 subunit, C825T polymorphism.
- Associated with an increased risk of hypertension, low plasma renin, & cardiac remodeling
- More prevalent in blacks (50%) than whites (10%)
- FH of DCM: female carriers of X-linked CM (e.g. Duchenne or Becker muscular dystrophy, Danon disease)
- Dietary-induced fluid retention (e.g. Northern Nigeria)
- Known association with anti-angiogenic signaling (a process exacerbated by preeclampsia)
- Definition - imprecise due to varied criteria across many international societies. 2010 European Society of Cardiology's listed below.
- (early) Pregnancy-associated CM (ePACM)
- Defined as CM occurring in 1st & 2nd trimesters
- Controversial
- Delineation may be arbitrary, as a study found no differences between PPCM and ePACM in the following characteristics:
- age, race, associated conditions, LVEF, the rate & time of recovery, maternal outcomes
- Defined as CM occurring in 1st & 2nd trimesters
- Peripartum cardiomyopathy (PPCM)
- Transplant rejection
- Myocardial depression can quickly develop and reverse with appropriate treatment
- Largely secondary to Lymphocytes, cytokines, and antibodies
- Eosinophilic myocarditis
- Toxic[2][3][4]
- Alcohol (>10% cases)
- Eti: 5-6 drinks (4 oz of pure EtOH) QS for 5-10years
- MOA: Direct toxicity of both Alcohol & its metabolite, acetaldehyde
- Genetic polymorphisms of genes encoding alcohol dehydrogenase & ACE make individuals at an increased risk of developing CM with prolonged alcohol exposure
- Complications:
- Early: Paroxysmal AFib "Holiday heart"
- Late: Persistent Afib, Withdrawal can worsen HF or arrhythmias Improvement can happen after 3-6months of abstinence
- Catecholamines: (Amphetamines, Cocaine, Pheochromocytoma, Stress-induced CM (Takotsubo's)
- MOA:
- Excess catecholamines can cause multifocal contraction band necrosis, likely secondary to calcium overload causing direct myocyte toxicity OR
- focal vasoconstriction in coronary artery in the setting of tachycardia, akin to ischemia-reperfusion with subsequent inflammation.
- often displays microinfarcts secondary to small vessel ischemia.
- Takotsubo's = older women after sudden intense emotional or physical stress + Global Ventricular dilatation with basal contraction
- Early animal studies suggest MOA: Intense sympathetic activation with heterogeneity of myocardial autonomic innervation, diffuse microvascular spasm, &/or direct catecholamine toxicity
- MOA:
- Chemotherapeutic agents - most common drugs causing Toxic CM
- Classical: Anthracyclines, Trastuzumab
- Anthracyclines
- RF for CM: Dose dependent (occurring once cumulative life-time dose >500mg/m2), Underlying cardiac disease, concomitant chest irradiation
- Figure 5, below, depicts the MOA of Anthracycline toxicity
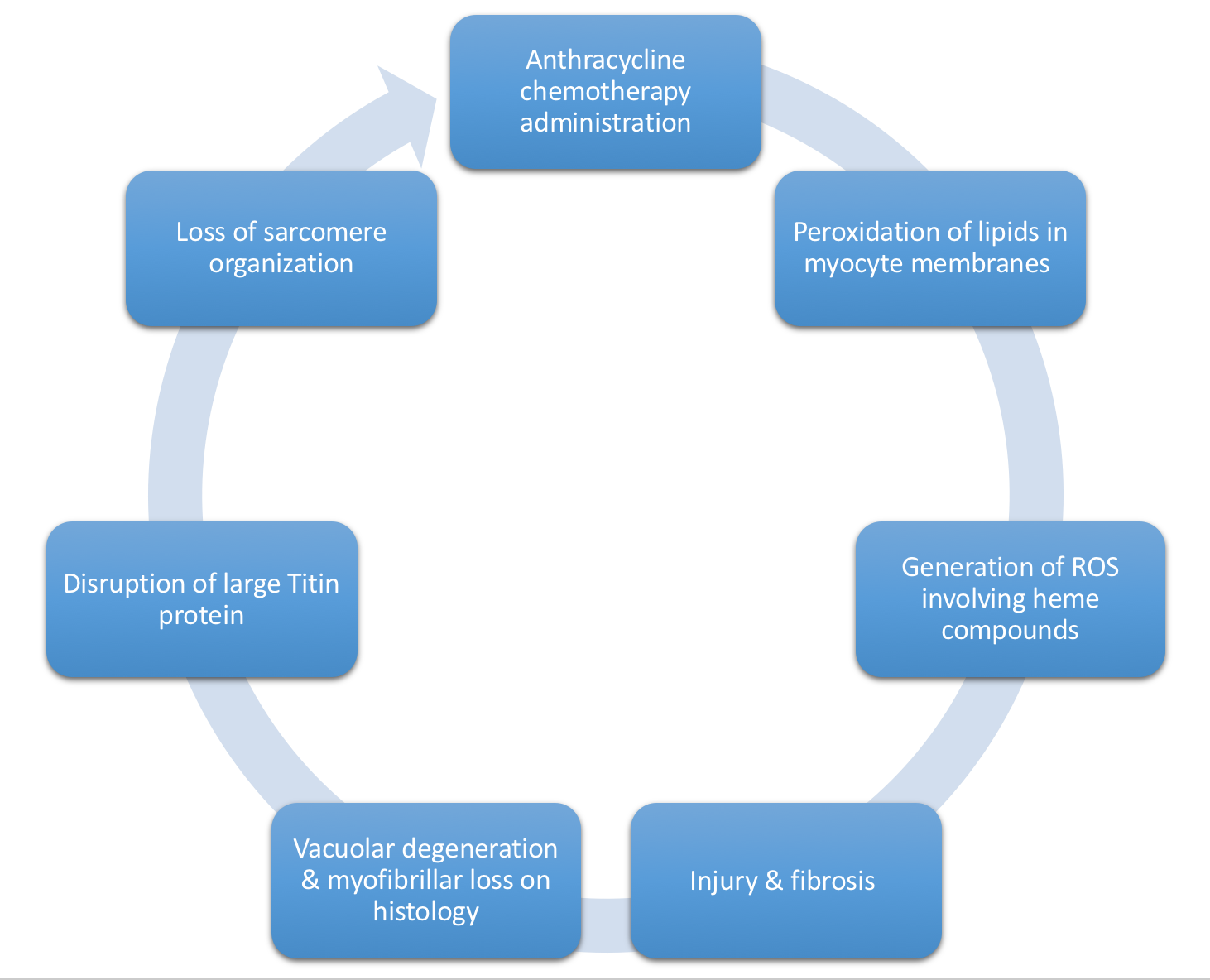
Figure 5. Mechanism of Anthracycline-induced dilated CM. ROS, Reactive Oxygen Species. - Generally, repeated cycles of the process shown above in Figure 5 → relatively nondilated ventricle secondary to underlying fibrosis → reduced EF (30-40%), unable to compensate by myocyte hypertrophy (due to underlying fibrosis).
- Time course varies from patient to patient.
- 3 different presentations of Anthracycline toxicity, grouped by chronology:
- Acute-onset HF (after single dose)
- Early-onset, dose-dependent anthracycline cardiotoxicity with resultant HF (during or shortly after a chronic course. Occurs in 3% of patients)
- A. Rapidly progressive
- B. Partial recovery of EF
- Chronic HF
- Presentation depends on the age the dose of anthracycline was received:
- A. Dose received before puberty: impaired development of the heart → clinical HF around age 20
- B. Dose received after puberty: gradual onset of HF Sx OR acute onset of HF following reversible 2nd hit/insult (e.g. influenza, Afib)
- Presentation depends on the age the dose of anthracycline was received:
- If managed appropriately, patient can live for years with compensated cardiac function
- Tyrosine kinase inhibitors (e.g. Traztuzumab, Imatinib)
- RF for CM: Concurrent use with anthracyclines
- MOA: Mab that interferes with cell surface receptors important for cardiac adaptation
- Usually CM is reversible, but not always.
- Some patients advance to HF and death
- Immune checkpoint inhibitors
- Anthracyclines
- Less common:
- Cyclophosphamide & Ifosfamide cardiotoxic in high doses.
- 5-FU, Cisplastin, & other Alkylating agents → coronary spasm → depressed contractility in some patients
- Immune checkpoint inhibitors
- Common examples:
- anti-CTLA-4 monoclonal antibodies (Ipilimumab, the original checkpoint inhibitor. Introduced in 2010)
- PD-1 monoclonal antibodies (eg, nivolumab, pembrolizumab)
- PD-L1 monoclonal antibodies (eg, atezolizumab)
- Can cause fatal myocarditis, HF, heart block, myocardial fibrosis and cardiomyopathy. Incidence <1%.
- Combination therapy with multiple checkpoint inhibitors has been associated with more severe and frequent myocarditis
- Possibly earlier cardiotoxicity, as well, compared to patients receiving a single checkpoint inhibitor (BMJ, NEJM)
- Combination therapy with multiple checkpoint inhibitors has been associated with more severe and frequent myocarditis
- Common examples:
- Classical: Anthracyclines, Trastuzumab
- IFN - can cause hypotension and arrhythmias.
- Other therapeutic agents (hydroxychloroquine, Chloroquine, Lithium, Phenothiazine antipsychotics, antiretroviral therapies)
- Drugs of misuse (emetine, anabolic steroids)
- Heavy metals: Lead, Mercury (amalgam fillings, cinnabar), Cobalt (tainted beer production)
- Occupational exposure: hydrocarbons, arsenicals
- Alcohol (>10% cases)
- Metabolic[2][3][4]
- Nutritional deficiencies: thiamine, selenium, carnitine. Responds to Rx.
- Beri-Beri
- Eti: Thiamine or Vitamin B1 deficiency
- Epi: Developing countries, EtOH, +/- teenagers consuming highly processed foods
- The importance of timely diagnosis is displayed below in Figure 6.

Figure 6. Patients with prolonged High-output HF will eventually lose systolic function without treatment, terminating in Low-output HF. This underscores the importance of prompt diagnosis and treatment.
- Selenium -- Keshan's disease
- Post bariatric surgery
- Beri-Beri
- Electrolyte deficiencies: calcium, phosphate, magnesium
- Calcium:
- Hypoparathyroidism (esp postsurgical)
- Intestinal dysfcn (diarrhea, s/p resection)
- Phosphorous
- Starvation
- Refeeding
- Magnesium = cofactor for B1-dependent reactions & for ATP
- Calcium:
- Endocrinopathy
- Thyroid disease
- Most common reason for thyroid abnormalities in patient with cardiac dyscrasias are treatment of tachyarrhythmias with amiodarone
- Hyperthyroidism and heart failure warrants very close inpatient monitoring, as decompensation may occur rapidly and have fatal consequences.
- Pheochromocytoma
- Heart failure with labile &/or orthostatic BP and episodic HTN
- DM
- Thyroid disease
- Obesity
- DM, HTN, Metabolic syndrome
- Independent fluid retention in obese people c impaired excretion (Rapid clearance of BNP by adipose tissue) → increase wall stress & secondary adaptive neurohormonal responsive.
- Hemochromatosis
- Primary/Hereditary
- 1 in 500 people
- AR form of HFE gene
- Low penetrance
- Secondary/Acquired
- Hemolytic anemia & transfusions
- MOA: excess iron deposited in the perinuclear compartment of cardiomyocytes, disrupting intracellular architecture and mitochondrial function
- Transferrin >60% : men :: Transferrin 45-50% : women
- Primary/Hereditary
- Nutritional deficiencies: thiamine, selenium, carnitine. Responds to Rx.
- Inherited Metabolic Pathway Defects[2][3][4]
- Familial (30%)
- MC mutation = TTN encoding titin (25% of familial disease)
- Men develop disease 1 decade b4 women.
- 8% Thick & thin filaments
- Ventricular Tachycardia & SCD a/w DESMOSOMAL proteins
- Atrial Arrhythmias, conduction disease, & CM : mutation LAMIN proteins :: Prominent FH of SCD or Vtach before clinical CM : Mutation of Desmosomal proteins
- Skeletal & Cardiac myopathy
- Dystrophin-related dystrophy (Duchenne's, Becker's) - X-linked
- Mitochondrial myopathies (e.g. Kearns-Sayre syndrome)
- Arrhythmogenic ventricular cardiomyopathy (AVC)
- Formerly arrhythmogenic right ventricular dysplasia (ARVD) or cardiomyopathy (ARVC), as first described in right ventricle.
- While has been described now in both ventricles, RV much more frequently affected
- Highly arrhythmogenic. Common cause of sudden cardiac death from malignant arrythmias (VTach, VFib)
- Pathophysiology depicted below in Figure 7.
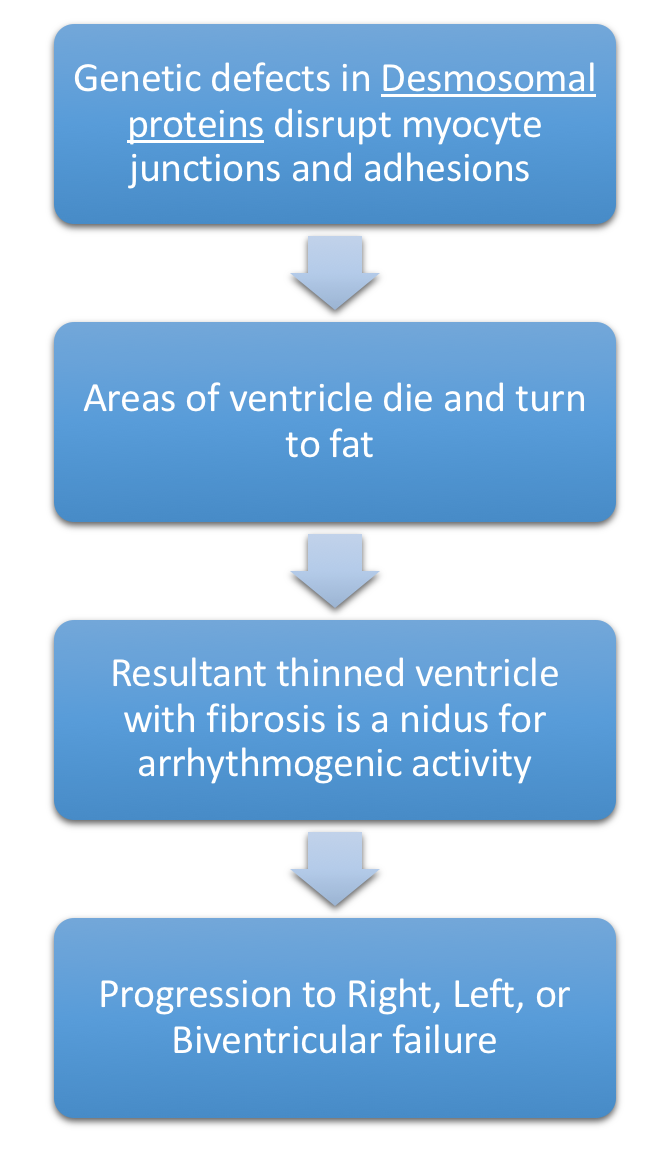
Figure 7. Pathophysiology of Arrhythmogenic Ventricular Dysplasia or Arrhythmogenic Ventricular Cardiomyopathy. Patients often report in Ventricular Tachycardia. Right ventricle is most commonly affected. - Genetic defects in Desmosomal proteins (especially plakoglobin & desmoplakin)
- Due to nonfunctional desmosomal proteins, patients have a distinctive phenotype, with striking woolly hair & thickened skin on palms & soles due to loss of elasticity in hair & skin
- Naxos syndrome = ARVC + hyperkeratosis of plantar palmar skin surfaces
- Formerly arrhythmogenic right ventricular dysplasia (ARVD) or cardiomyopathy (ARVC), as first described in right ventricle.
- Hemochromatosis
- Associated with other systemic disease
- Susceptibility to immune-mediate myocarditis
- Overlap with Nondilated Cardiomyopathy
- "Minimally dilated CM"
- Hemochromatosis
- Amyloidosis
- Hypertrophic CM
- Familial (30%)
- Miscellaneous (Shared Elements of Above Etiologies)[2][3][4]
- Arrhythmogenic Ventricular (RV>LV) dysplasia
- LV Noncompaction (LVNC) [3]
- Three cardinal features = Ventricular arrhythmias + Embolic events + HF
- Pathophysiology not fully characterized, but evolving. Involves interaction with genetics and environment (variable penetrance):
- Gross pathology: Multiple trabeculations in LV distal to papillary MM → spongy appearance of the apex
- Microscopic pathophysiology: Associated with mutations in Sarcomeric & Tafazzin genes
- Tafazzin gene encodes a protein expressed at high levels in cardiac & skeletal muscle.
- Mutations in this gene have been associated with a number of cardiomyopathies.
- Sarcomeric genes identified associated with mutations causing DCM & HCM (e.g. MYH7, MYBPC3, TTN)
- Study by van Waning et al. highlights the expanding role of genetics
- Genetic classification
- 1) Identified genetic mutations (32%)
- More often children
- Multiple mutations & TTN mutations (particularly TTN A-band truncation mutations) were highest risk for LV systolic dysfunction & major adverse cardiac events (MACE)
- 2) Probably genetic etiology (family history of CM, but no identified mutation) (16%)
- 3) Sporadic cases (52%)
- Commonly adults)
- Index mutation can produce different phenotypes in relatives
- Asymptomatic
- Usually represent mutations on several domains on TTN gene (e.g. Z-disc, I-band, M-band) rather than just A-band mutations
- DCM
- MYH7 and TTN A-band mutations most commonly associated with DCM
- HCM
- MYBPC3 gene mutations most commonly associated with HCM
- Asymptomatic
- Focal truncation mutation primarily in TTN A-band : DCM phenotype :: Uniformly distributed truncation mutation (Z-disc, M/I/A bands of TTN) : Asymptomatic phenotype
- 1) Identified genetic mutations (32%)
- Phenotypic classification
- 3 subtypes
- 1) Isolated LVNC (asymptomatic)
- 2) LVNC + DCM
- Highest risk of systolic dysfunction, MACE, DCM without LVNC in relatives
- 3) LVNC + HCM
- 3 subtypes
- Genetic classification
- Tafazzin gene encodes a protein expressed at high levels in cardiac & skeletal muscle.
- Diagnosis usually made on imaging
- Jenni echocardiographic criteria most frequently used.
- Based on ratio between severely thickened myocardium with noncompacted layer ≥ 2x as thick as compacted layer (measured in short axis view)
- Jenni echocardiographic criteria most frequently used.
- Peripartum CM (see details above)
- Tachycardia-related cardiomyopathy
- SVT arrhythmias with uncontrolled rate
- Very frequent nonsustained Ventricular tachycardia or High PVC burden
- LBBB



HYPERTROPHIC CARDIOMYOPATHY
[edit | edit source]Hypertrophic Cardiomyopathy with or without obstruction is characterized by a thickened, hypertrophic left ventricular wall, with hyperdynamic cardiac function, and no associated hemodynamic factors (HTN, Aortic valve disease, Systemic infiltrative/storage disease).
Epidemiology and Genetics[2][3][4]
[edit | edit source]- Disease first described in 1957.
- Prevalence 1:500 (~Hereditary hemochromatosis)
- Risk of Sudden Cardiac Death in patients with HCM is 0.5%
- Transmitted in an Autosomal Dominant pattern
- HCM is age dependent, with incomplete penetrance
- Patients developing disease later in life have fare better than those with disease in adolescence/young adulthood.
- Same mutation in related individuals can produce different phenotype
- Various patterns of pattern of hypertrophy concentric vs. asymmetric, presence or absence of outflow obstruction, development of Afib or malignant arrhythmia, SCD)
- A majority of the work on the genetics of cardiomyopathy was initially completed in this population
- With advances in genetics, linkage analyses identified sarcomeric mutations in 60% of patients with HCM.
- >1400 mostly missense mutations in 9 different genes
- Rates of sarcomeric mutations exceed 60% in patients with familial disease & asymmetric septal hypertrophy.
- 80% of HCM patients have a mutation in either Myosin-binding protein C (MYBPC3) or ß-myosin heavy chain (MHY7) gene loci encoding sarcomeric proteins[5][6]
- MYBPC3 is the most commonly mutated gene in HCM.
- MYBPC3 > MHY7 (sarcomeric) gene mutations : Most common mutations in HCM :: V122I : Most common mutation in Amyloidosis, especially African Americans
- With advances in genetics, linkage analyses identified sarcomeric mutations in 60% of patients with HCM.
Microscopic pathophysiology[2][3][4]
[edit | edit source]- Pathophysiology of HCM can be summarized in three aspects:
- 1) Fibrosis
- secondary to early activation of profibrotic pathways
- Interstitial fibrosis detectable before overt hypertrophy
- Focal areas of 'replacement fibrosis' detectable on MRI before hypertrophy present
- Myocardial fibroblast exaggerated response to primary myocardial dysfunction
- Areas of scarring may represent substrate for malignant ventricular arrhythmias (MCC of death in this population)
- Focal areas of 'replacement fibrosis' detectable on MRI before hypertrophy present
- Over time, fibrosis → diastolic dysfunction
- 2) Disorganized hypertrophy (predominant)
- 3) Microvascular disease
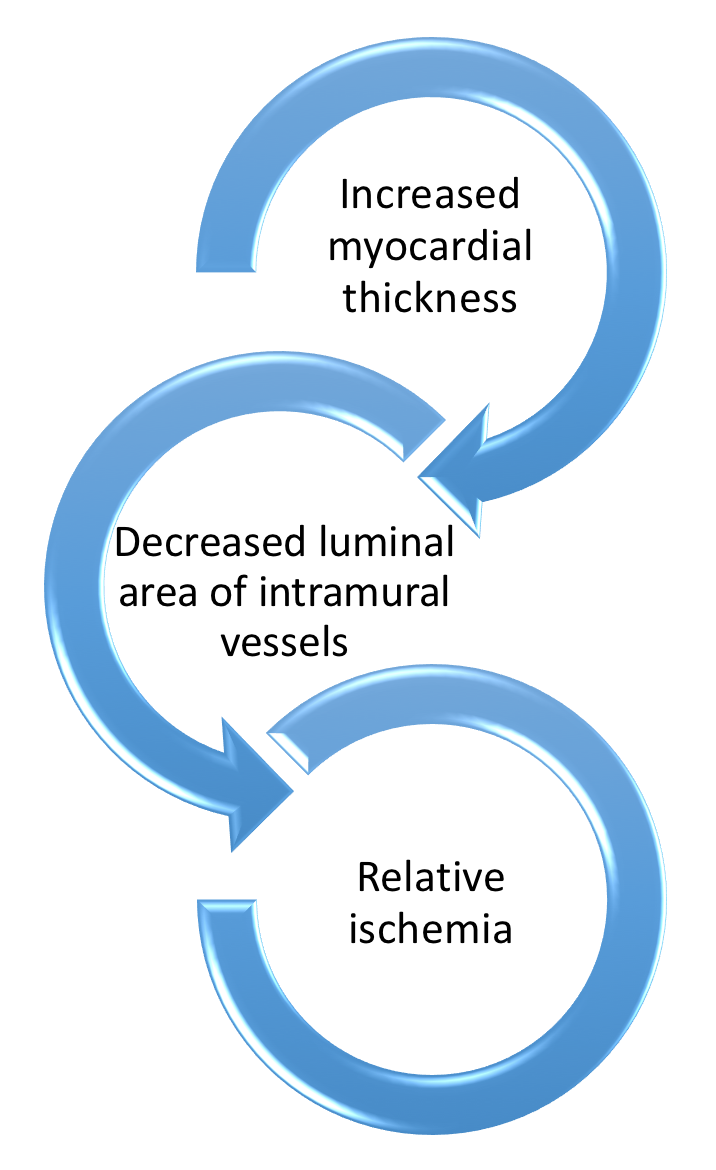
Figure 8. HCM pathophysiology of microvascular disease over time. - Microinfarction of hypertrophied myocardium proposed mechanism for replacement scar formation
- 1) Fibrosis
- On the individual sarcomere level, HCM mutations produce modification in regulatory proteins leading to:
- Enhanced calcium sensitivity
- Maximal force generation
- Increased ATPase activity
- This changes on a microscopic level lead to disorganized hypertrophy, causing a cycle of:
- Abnormal energetics
- Impaired relaxation

Macroscopic pathophysiology[2][3][4]
[edit | edit source]- Nonuniform ventricular thickening
- Location of maximal hypertrophy may vary:
- IV Septum (MC)
- IV Septum : LV free wall thickness ratio of 3:1
- Concentric
- Midventricular
- Apex (Yamaguchi's syndrome)
- Less commonly familial
- Only 15% derived from sarcomeric mutations
- IV Septum (MC)
- Fibrosis, disorganized hypertrophy, and microvascular disease contribute to diastolic & contractile dysfunction along with elevated intracardiac pressures → higher wall stress & Myocardial O2 demand
- LV outflow tract (LVOT) Obstruction is present in 30% of patients at rest, 30% inducible by exercise
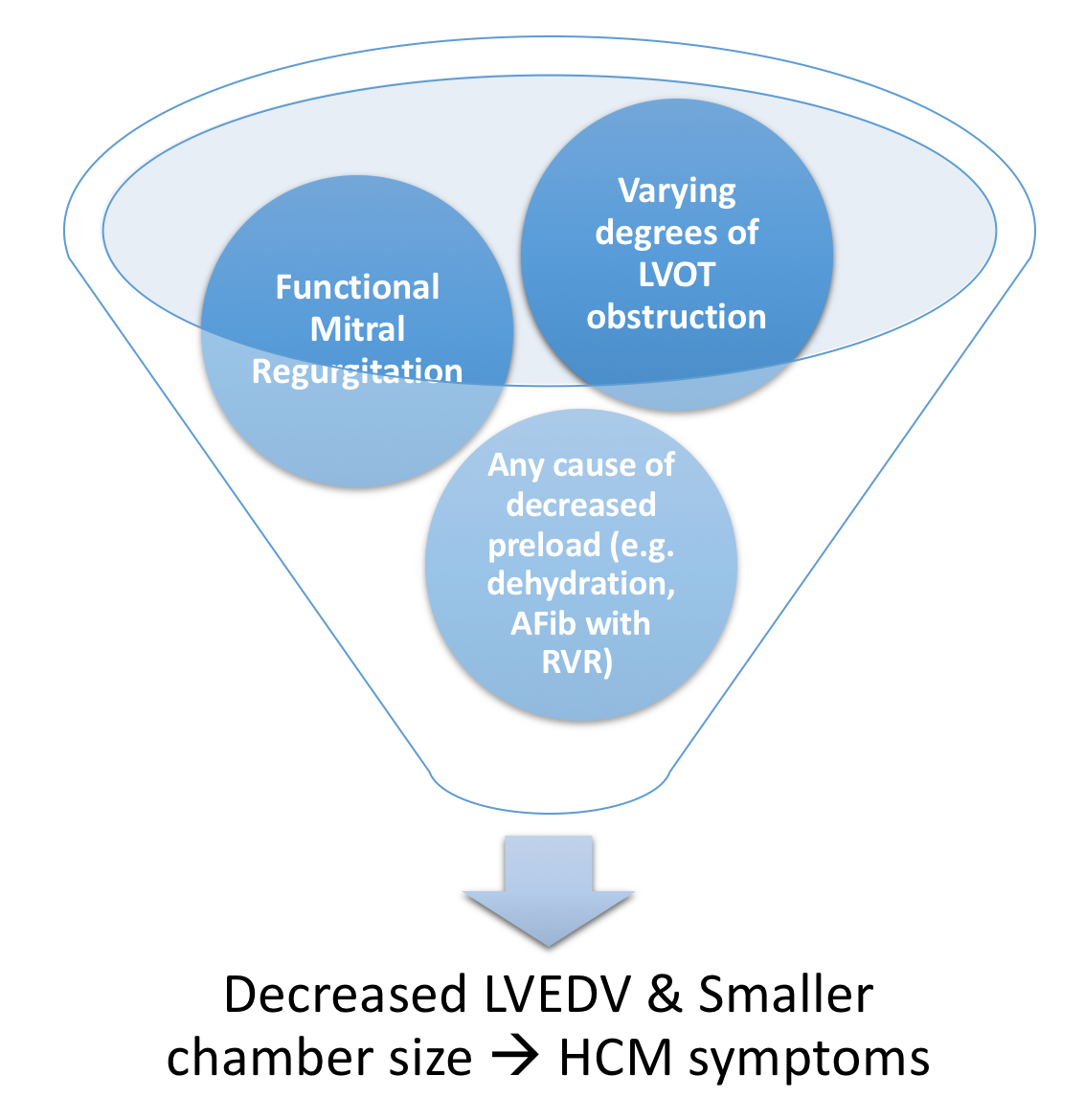
Figure 9. Pathophysiology of LVOT obstruction in HCM - Varying degrees of LVOT obstruction is the primary mechanism of HCM
- This occurs via drag forces push the anterior mitral leaflet in contact with hypertrophied ventricular septum
- Compared to individuals without HCM, the anterior mitral leaflet is anteriorly displaced & thickened from fibrous endocardial plaque deposition)
- Functional Mitral Regurgitation occurs as the anteriorly displaced MV may cause a posteriorly directed regurgitant jet
- Decreased Left Ventricular End Diastolic Volume LVEDV
- To maintain SV across LVOT obstruction :
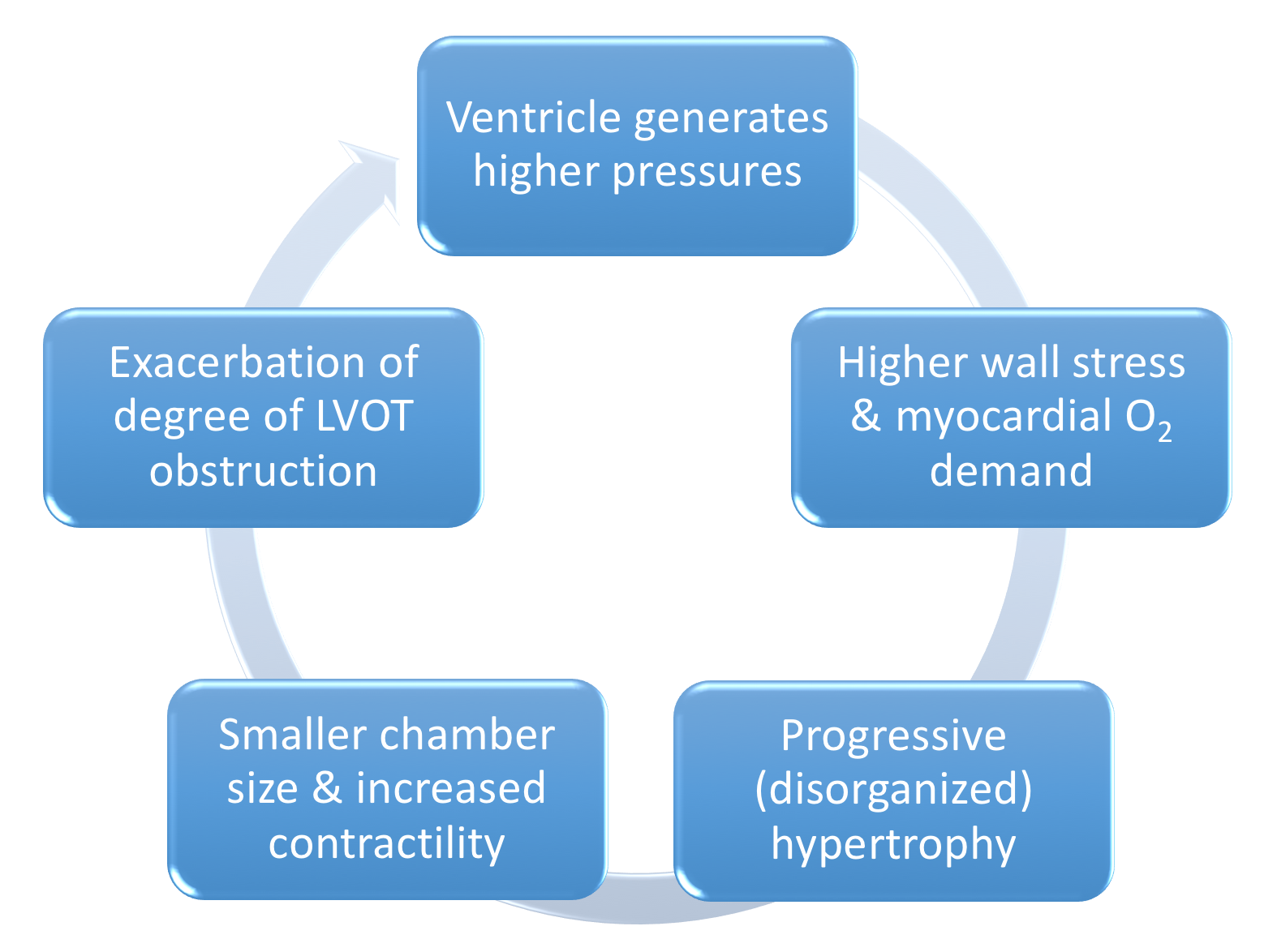
Figure 10. Pathological cycle of LVOT obstruction in HCM - Presyncope & hypotension can result from decreased preload (e.g. diuretics) or decreased afterload (e.g. arterial dilators)
- Risk of Sudden Cardiac Death in patients with HCM is 0.5% [3]
- Factors increasing risk of SCD in HCM - PS FELA:
- Prior Cardiac arrest or Spontaneous sustained VTach
- Spontaneous NSVT
- FH of SCD
- Exertional or nonvagal syncope
- LV thickness >30mm
- Abnormal BP response to exercise (failure to increase SBP to ≥120mmHg OR decline in SBP during exercise)
- Factors increasing risk of SCD in HCM - PS FELA:
- Afib is common in patients with HCM. RVR is poorly tolerated (decreased preload, decreased filling, increased myocardial O2 demand). Following modalities used for treatment:
- AV nodal (AVN) agents:
- Beta-blockers, Non-dihydropyridine-CCB (verapamil, diltiazem)
- Rate control may be necessary to sustain atrial kick - Amiodarone, Disopyramide
- If refractory to medical therapy: AVN Ablation
- AV nodal (AVN) agents:
- HCM vs. Athlete's heart: Athlete's heart ventricular hypertrophy regresses with cessation of training.
- Also, patient's have supranormal exercise capacity, mild ventricular dilation, and normal diastolic function


RESTRICTIVE CARDIOMYOPATHY
[edit | edit source]Restrictive CM is the least common CM phenotype. [2] [3] Figure 11 below contrasts Restrictive CM with the two more common etiologies.

Restrictive CM is characterized by the following 6 characteristics:[2][3]
- Abnormal diastolic function
- Mildly decreased contractility & EF (usually >30-50%)
- (often massive) Biatrial enlargement
- Modest LV dilation (LVEDV <6cm)
- RVEDP & LVEDP both increased
- Preservation of CO until late in the disease.
The interactive pathophysiology of Restrictive CM is depicted below in Figure 12:

Common causes of restrictive CM are categorized below.
| Subtype | Examples |
|---|---|
| Infiltrative (between myocytes) | Amyloidosis, Inherited metabolic defects |
| Storage (within myocytes) | Hemochromatosis, Inherited metabolic defects (Fabry's, Gaucher's) |
| Fibrotic | Radiation (breast/lung CA, Mediastinal lymphoma)
Scleroderma Anthracyclines |
| Endomyocardial | Possibly related fibrotic disease (Tropical endomyocardial fibrosis, Hypereosinophilic syndrome (Löffler's endocarditis))
Carcinoid syndrome Radiation Medication |
| Overlap with Other Cardiomyopathies | Hypertrophic cardiomyopathy/"pseudohypertrophic"
"Minimally dilated" cardiomyopathy Sarcoidosis |
| Idiopathic | ----- |
Detailed Causes of Restrictive CM[2][3][4][7]
- Infiltrative disorders (between myocytes)
- Amyloidosis
- several proteins can fold into Beta-pleated sheets, causing phenotypic variability.
- >100 identified mutations in Transthyretin on chromosome 13 (locus heterogeneity).
- MOA of organ dysfunction twofold:
- physical disruption from infiltrating amyloid fibrils
- direct toxicity from immunoglobulin light chain & abnormal transthyretin protein aggregates.
- Subtypes listed below in order of decreasing severity.
- Primary (light chain amyloid)
- Cardiac Sx
- Neuro Sx
- Familial (abnormal transthyretin)
- Cardiac Sx
- Neuro Sx
- V122I transthyretin mutation is present in 10% of African Americans with heart failure and approximately ~4% of the African Americans population.
- V122I gene mutation : Amyloidosis & Heart Failure :: BRCA1/2 gene mutation : Breast CA
- Senile (abnormal accumulation (quantity) of normal transthyretin or atrial peptides)
- 10% of people >80yo & 50% of people >90yo
- Increasing number
- Often asymptomatic. Men more likely to display symptoms as they have a higher burden disease.
- Primary (light chain amyloid)
- Inherited metabolic defects
- Amyloidosis
- Storage disorders (within myocytes)
- Hemochromatosis (iron)
- Inherited metabolic defects - Enzyme replacement may help
- Fabry's disease (Alpha-galactosidase A deficiency)
- Lysosomal enzyme
- X-linked. Caused by one of >160 mutations (Allelic heterogeneity)
- Lamellar inclusions of glycosphingolipids
- Glycogen storage disease (II, III)
- Most are diagnosed early in childhood & die before adulthood
- Fabry's disease (Alpha-galactosidase A deficiency)
- Fibrotic - Restrictive myocardial disease without ventricular dilation
- Radiation (breast/lung CA, Mediastinal lymphoma)
- Fibrotic restrictive CM can coexist with constrictive pericarditis
- Pericardiectomy does not work well in this setting.
- Scleroderma
- Small vessel spasm and ischemia → small, stiff heart with rEF without dilation
- Concomitant Pulmonary HTN accentuates Right HF present in most restrictive diseases
- Anthracyclines
- Primarily a dilated CM with restrictive component
- Fibrosis limits the degree of dilation possible
- Radiation (breast/lung CA, Mediastinal lymphoma)
- Endomyocardial
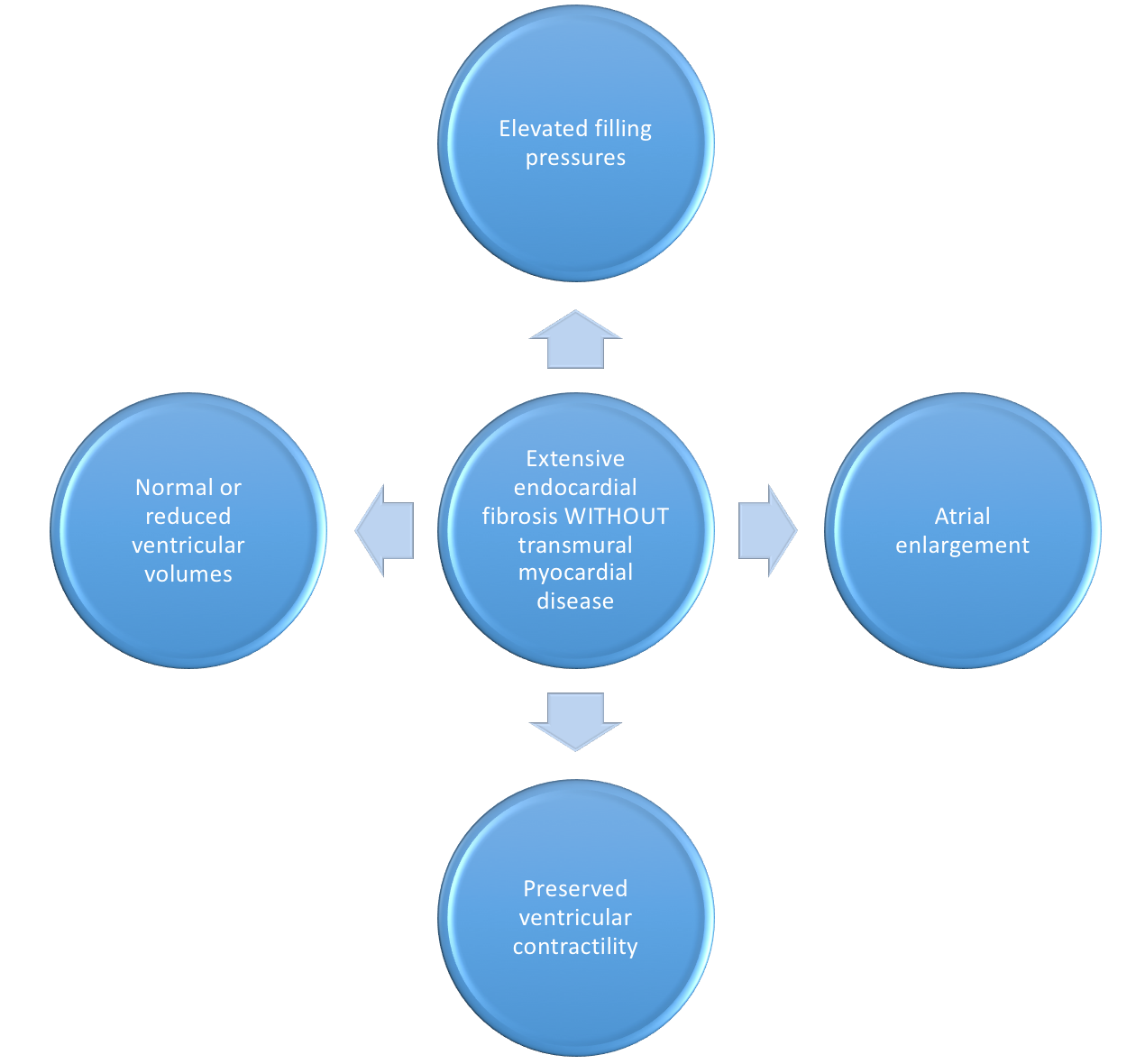
Figure 13. Pathophysiology of Endocardial fibrosis and it's ensuing symptomatic presentation. - Possibly related fibrotic disease
- Tropical endomyocardial fibrosis[3][7]
- Most common cause of Endomyocardial fibrosis near the equator
- Found in areas
- May comprise up to 25% of CHF in these regions
- More common in African Americans
- MOA:
- Unclear. Leading theories include:
- End stage of prior hypereosinophilic disease triggered by endemic parasites
- Regional nutritional deficiencies
- Unclear. Leading theories include:
- Fibrosis obliterating the ventricular apex, extending to valvular apparatus, like Löffler's endocarditis
- Associated with pericardial effusions, unlike Löffler's endocarditis
- Fibrosis obliterating the ventricular apex, extending to valvular apparatus : Tropical endomyocardial fibrosis AND Löffler's endocarditis :: Associated with pericardial effusions : Tropical endomyocardial fibrosis NOT Löffler's endocarditis
- Hypereosinophilic syndrome (Löffler's endocarditis)[3][7]
- Most common cause of Endomyocardial fibrosis outside of equatorial regions
- Men > Women
- Hypereosinophilia >1500eos/mm3 for ≥6mo
- Hypereosinophilia >1500eos/mm3 for ≥6mo : Hypereosinophilic syndrome (Löffler's endocarditis) :: WBC 15, Cr 1.5 : Severe C. Diff
- Etiology often unclear (idiopathic)
- CHINA is a mnemonic for chronic causes of eosinophilia. Helminth, Allergic, Neoplastic most common identifiable causes
- Connective Tissue disease (e.g. Eosinophilic granulomatosis with Polyangiitis EGP)
- Helminthic/Parasitic (esp. Strongyloides)
- Idiopathic hypereosinophilic syndrome
- Neoplastic - (lymphomas, esp Hodgkin's, CML)
- Hypereosinophilic syndrome associated with myeloproliferative disorders are often secondary to chromosomal rearrangements involving platelet-derived growth factor receptor (PDGFR), creating a fusion gene yielding a constitutively active PDGFR tyrosine kinase
- Treatment with Imatinib (TKI) has produced hematologic remissions and reversal of endomyocarditis
- Allergy/Atopy/Asthma/phArmAceuticAl-induced eosinophilia (i.e. DRESS from CArbAmAzepine, SulfonAmides)
- HAN : most common (MC) identifiable cause of Eosinophilia :: CHINA : most common cause of (MCC) eosinophilia
- 2 phases of eosinophilic myocardial disease:
- Acute inflammatory phase
- Eosinophils damage the endocardium
- Systemic injury to other organs
- Subacute/chronic fibrotic phase
- Cardiac inflammation replaced by fibrosis with superimposed thrombosis
- In severe disease, the fibrotic layer can:
- obliterate the ventricular apex
- extend to the AV valve leaflets causing fibrotic, thickened valvular apparatus
- In severe disease, the fibrotic layer can:
- Unclear risk factors or triggers to mark the transition from hypereosinophilic syndrome to extensive fibrosis
- Symptoms: CHF (Right>Left), Embolic events, Atrial arrhythmias
- Cardiac inflammation replaced by fibrosis with superimposed thrombosis
- Acute inflammatory phase
- Most common cause of Endomyocardial fibrosis outside of equatorial regions
- Tropical endomyocardial fibrosis[3][7]
- Carcinoid syndrome
- Only occurs with liver mets, as liver unable to metabolize serotonin → more serotonin released to venous circulation → Systemic Serotonin → fibrous plaques in the endocardium & heart valves (Right >>> Left)
- Associated symptoms of flushing & wheezing
- Only occurs with liver mets, as liver unable to metabolize serotonin → more serotonin released to venous circulation → Systemic Serotonin → fibrous plaques in the endocardium & heart valves (Right >>> Left)
- Radiation
- Medications: e.g. Serotonin, Ergotamine
- Overlap with Other Cardiomyopathies
- Hypertrophic cardiomyopathy/"pseudohypertrophic"
- Pseudohypertrophy refers to thickened myocardium secondary to infiltration of products between or within cells
- Restrictive phenotype predominates, but may also concurrently have mildly dilated CM
- "Minimally dilated" cardiomyopathy
- Early-stage dilated cardiomyopathy
- Partial recovery from dilated cardiomyopathy
- Sarcoidosis
- Hypertrophic cardiomyopathy/"pseudohypertrophic"
- Idiopathic

Gross Pathology
[edit | edit source]-
 Cardiomyopathy: Gross excellent view of mitral valve from left atrium anterior leaflet appears to balloon a bit into the atrium
Cardiomyopathy: Gross excellent view of mitral valve from left atrium anterior leaflet appears to balloon a bit into the atrium -
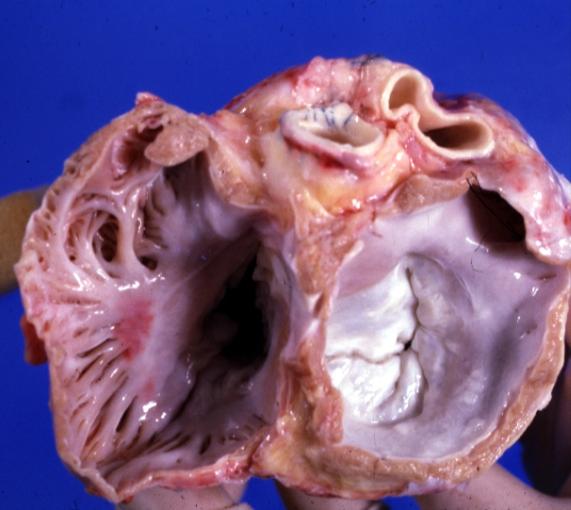 Cardiomyopathy: Gross excellent view of mitral and tricuspid valves from atria, appear normal anatomy.
Cardiomyopathy: Gross excellent view of mitral and tricuspid valves from atria, appear normal anatomy.


-
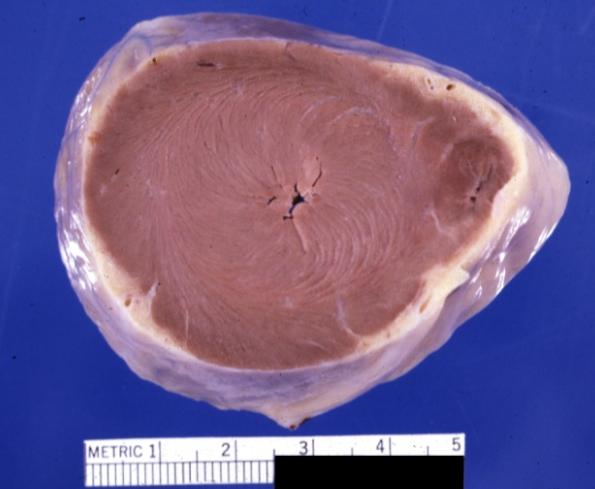 Cardiomyopathy: Gross apical slice of left and right ventricles concentric hypertrophy with cavitary obliteration sudden unexpected death obstructive cardiomyopathy
Cardiomyopathy: Gross apical slice of left and right ventricles concentric hypertrophy with cavitary obliteration sudden unexpected death obstructive cardiomyopathy -
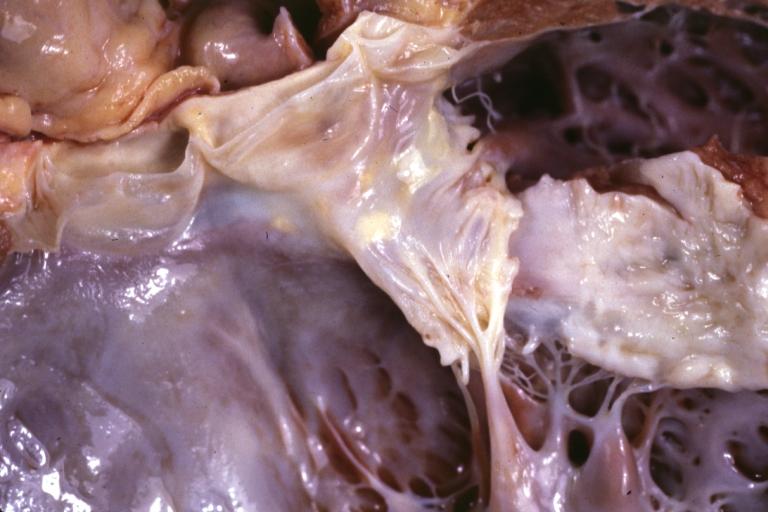 Dilated Cardiomyopathy: Gross natural color close-up view of heart surgically removed for a transplantation shows aortic valve and anterior leaflet of mitral valve with cholesterol deposits endocardium of left ventricle is diffusely thickened
Dilated Cardiomyopathy: Gross natural color close-up view of heart surgically removed for a transplantation shows aortic valve and anterior leaflet of mitral valve with cholesterol deposits endocardium of left ventricle is diffusely thickened


-
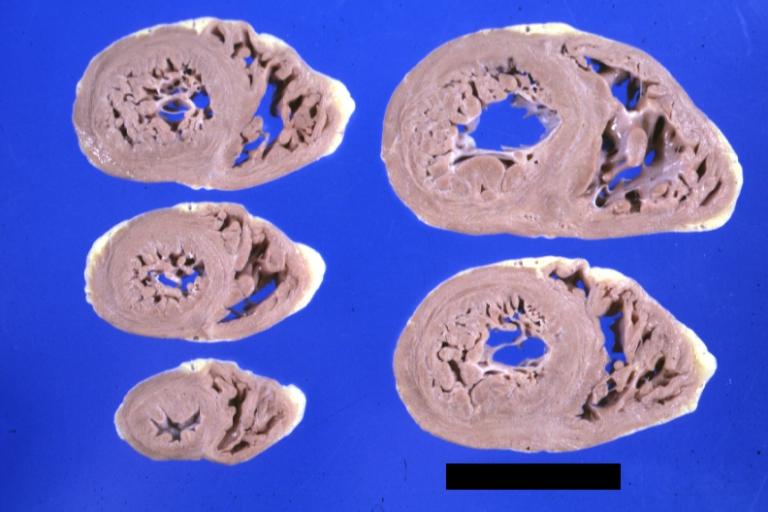 Cardiomyopathy: Gross montage of ventricular slices showing hypertrophy and about normal ventricular lumen size a hypertrophic non-dilated cardiomyopathy
Cardiomyopathy: Gross montage of ventricular slices showing hypertrophy and about normal ventricular lumen size a hypertrophic non-dilated cardiomyopathy -
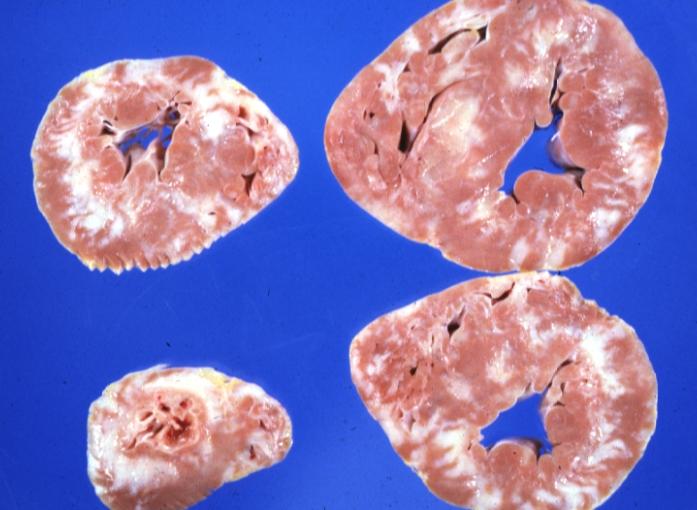 Cardiomyopathy: Gross ventricular slices hypertrophy and extensive myocardial fibrosis a unique case of global fiber disarray with atrophy and fibrosis
Cardiomyopathy: Gross ventricular slices hypertrophy and extensive myocardial fibrosis a unique case of global fiber disarray with atrophy and fibrosis


-
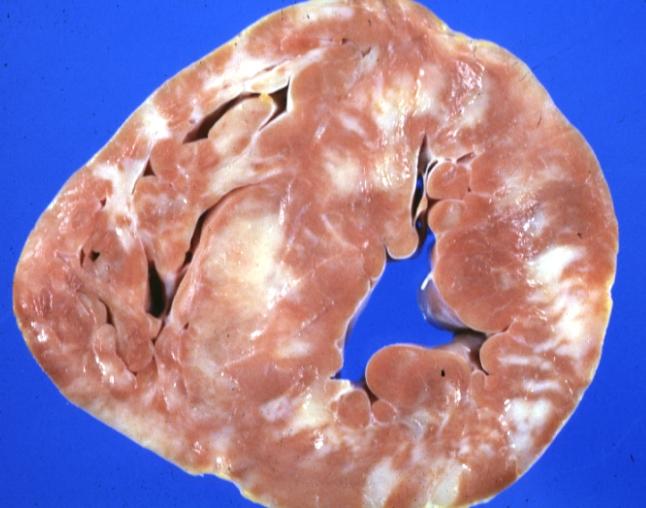
 Cardiomyopathy: Gross close up view of a ventricle slice
Cardiomyopathy: Gross close up view of a ventricle slice -
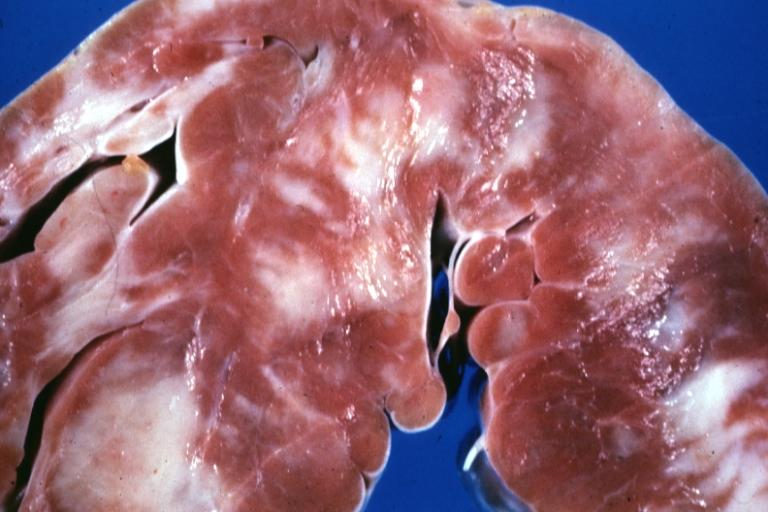
 Cardiomyopathy: Gross excellent ventricular slice with hypertrophy and fibrosis a unique case of global fiber disarray with hypertrophy then atrophy and then fibrosis
Cardiomyopathy: Gross excellent ventricular slice with hypertrophy and fibrosis a unique case of global fiber disarray with hypertrophy then atrophy and then fibrosis


-
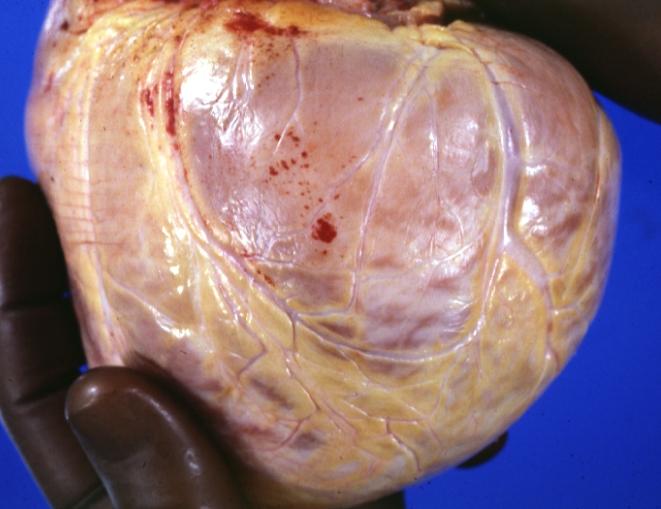 Cardiomyopathy: Gross external view of globular heart with patchy fibrosis seen through epicardium
Cardiomyopathy: Gross external view of globular heart with patchy fibrosis seen through epicardium -
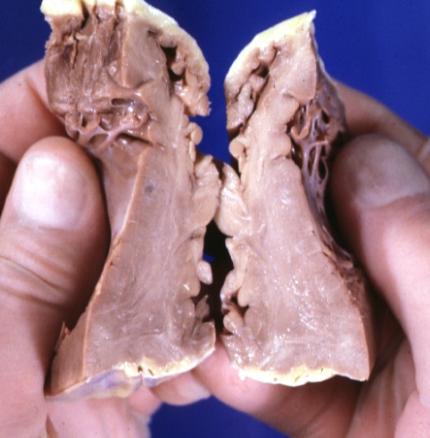 Cardiomyopathy: Gross interventricular septum showing asymmetrical hypertrophy in posterior septum
Cardiomyopathy: Gross interventricular septum showing asymmetrical hypertrophy in posterior septum


-
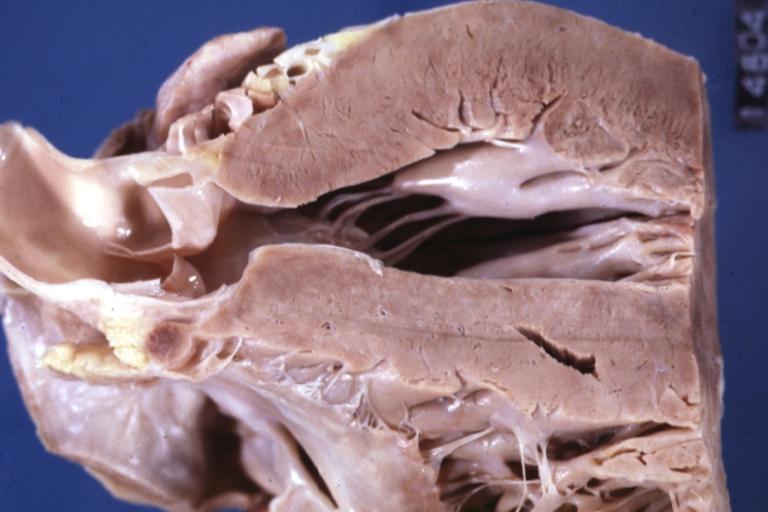 Cardiomyopathy: Gross hypertrophic cardiomyopathy obstructive excellent section through left ventricle outlet to show subvalvular narrowing case of sudden death in a 27 yo male playing basketball no history of disease
Cardiomyopathy: Gross hypertrophic cardiomyopathy obstructive excellent section through left ventricle outlet to show subvalvular narrowing case of sudden death in a 27 yo male playing basketball no history of disease -
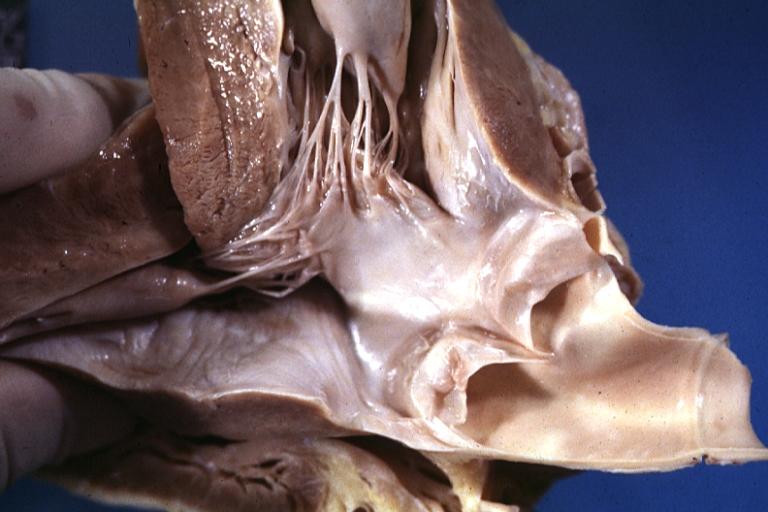 Cardiomyopathy: Gross obstructive cardiomyopathy showing aorta outflow tract with marked endocardial thickening mitral valve appears normal (Same case as previous one)
Cardiomyopathy: Gross obstructive cardiomyopathy showing aorta outflow tract with marked endocardial thickening mitral valve appears normal (Same case as previous one)


-
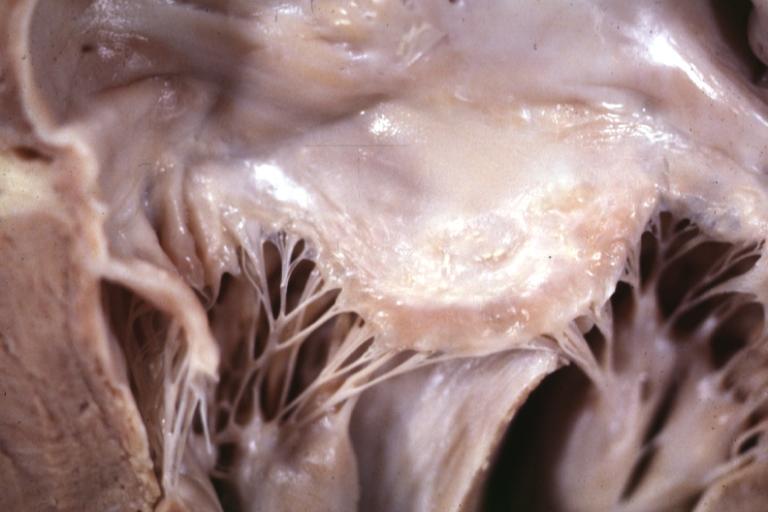 Cardiomyopathy: Gross excellent view of mitral valve atrial surface showing thickening which is fibrous in body of valve and myxoid at area of free margin changes presumed secondary to insufficiency due to anterior motion
Cardiomyopathy: Gross excellent view of mitral valve atrial surface showing thickening which is fibrous in body of valve and myxoid at area of free margin changes presumed secondary to insufficiency due to anterior motion -
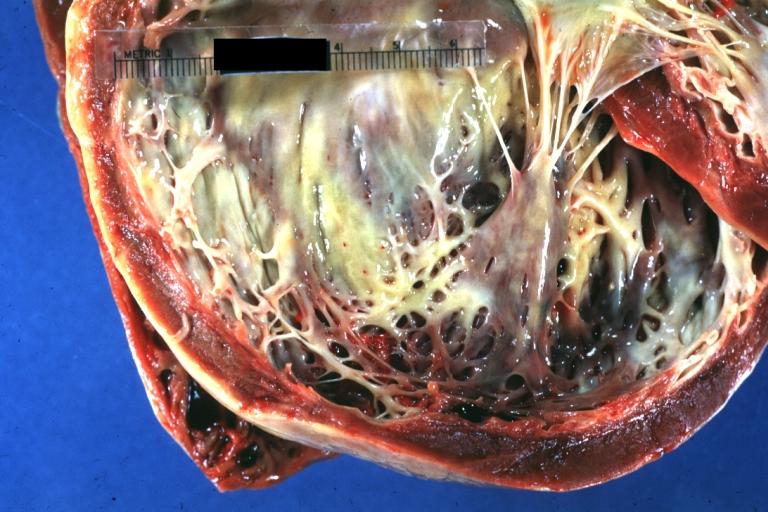 Cardiomyopathy: Gross dilated left ventricle with marked endocardial thickening this is what has been called adult fibroelastosis
Cardiomyopathy: Gross dilated left ventricle with marked endocardial thickening this is what has been called adult fibroelastosis


-
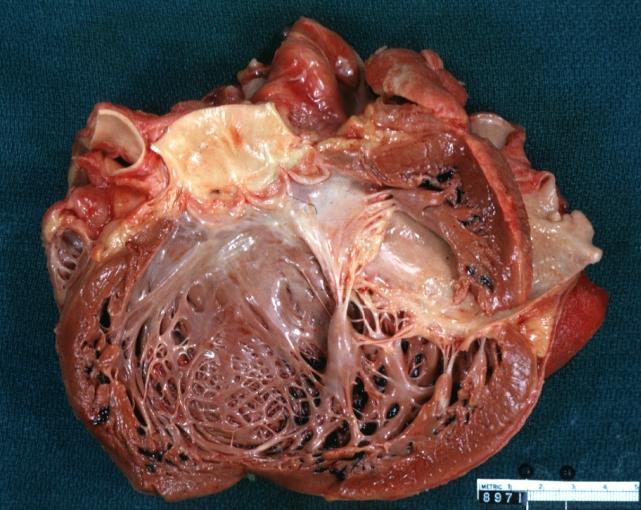 Dilated Cardiomyopathy: Gross good example huge dilated left ventricle
Dilated Cardiomyopathy: Gross good example huge dilated left ventricle -
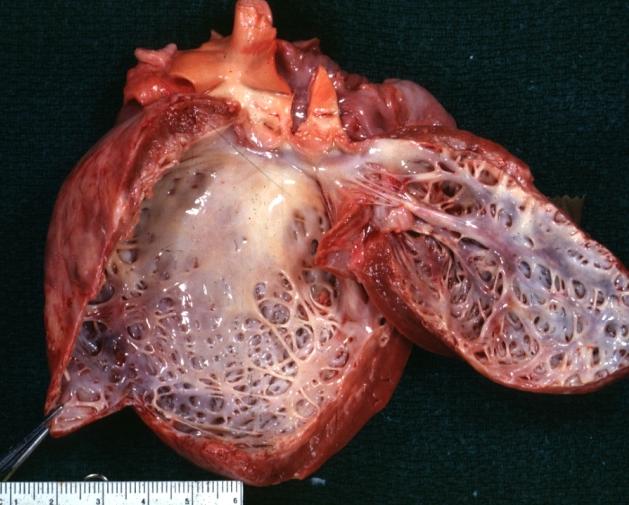 Dilated Cardiomyopathy: Gross dilated left ventricle with marked endocardial sclerosis (an excellent example)
Dilated Cardiomyopathy: Gross dilated left ventricle with marked endocardial sclerosis (an excellent example)


-
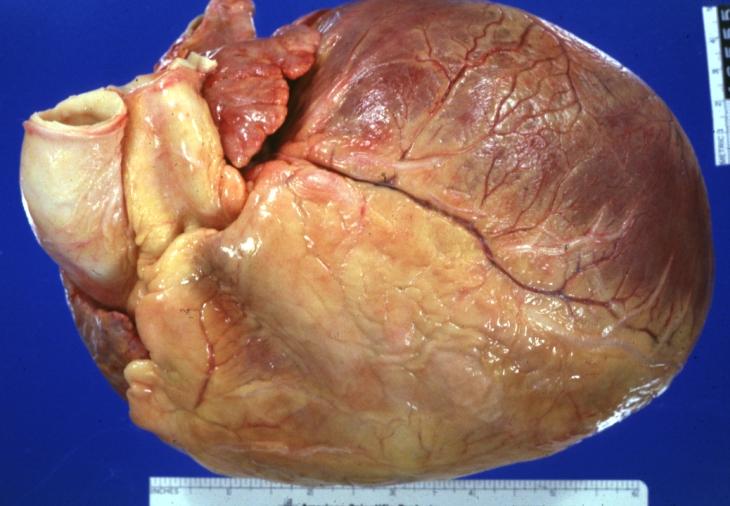 Cardiomyopathy: Gross intact globular shaped heart
Cardiomyopathy: Gross intact globular shaped heart -
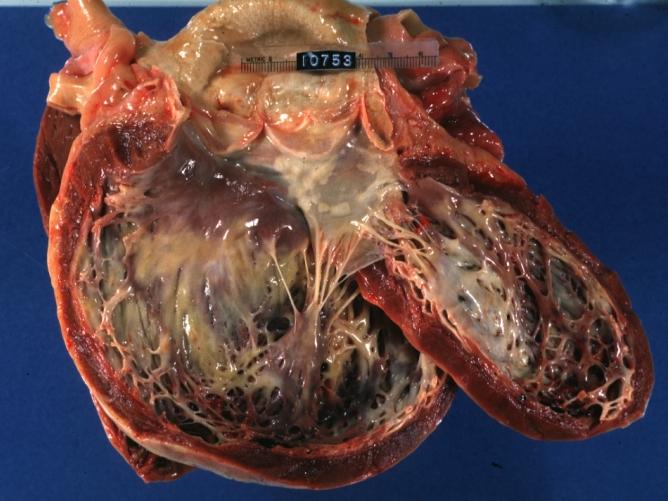 Dilated Cardiomyopathy: Gross opened left ventricle dilated with endocardial thickening good example
Dilated Cardiomyopathy: Gross opened left ventricle dilated with endocardial thickening good example


-
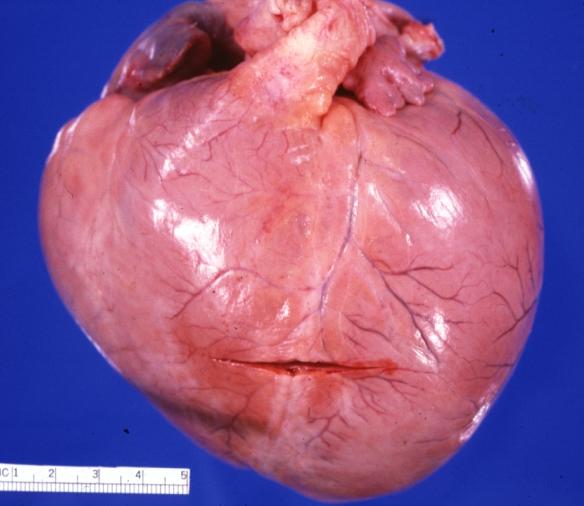 Cardiomyopathy: Gross globular heart external view 10 year old girl with sickle cell anemia
Cardiomyopathy: Gross globular heart external view 10 year old girl with sickle cell anemia -
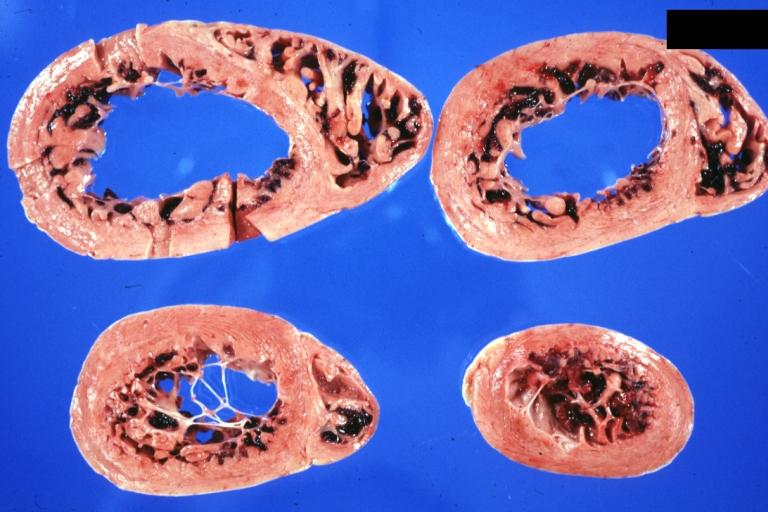 Cardiomyopathy: Gross horizontal sections of ventricles dilation type 10 year old girl with sickle cell anemia
Cardiomyopathy: Gross horizontal sections of ventricles dilation type 10 year old girl with sickle cell anemia


-
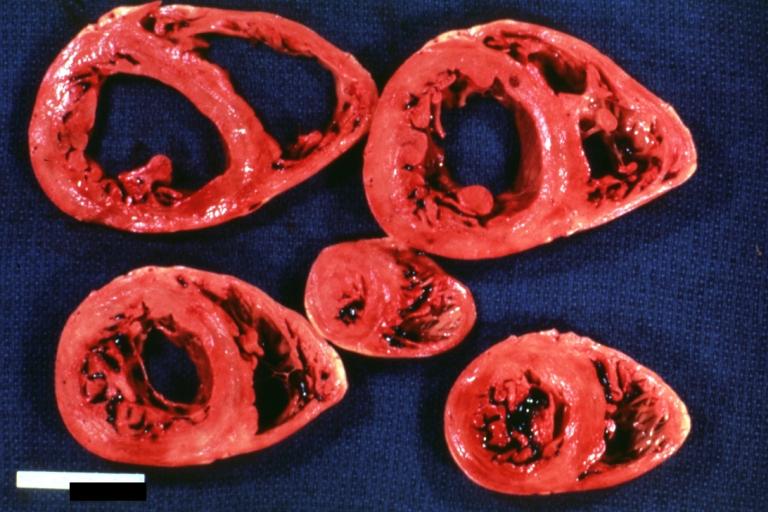 Cardiomyopathy: Intermediate between hypertrophic and dilated
Cardiomyopathy: Intermediate between hypertrophic and dilated -
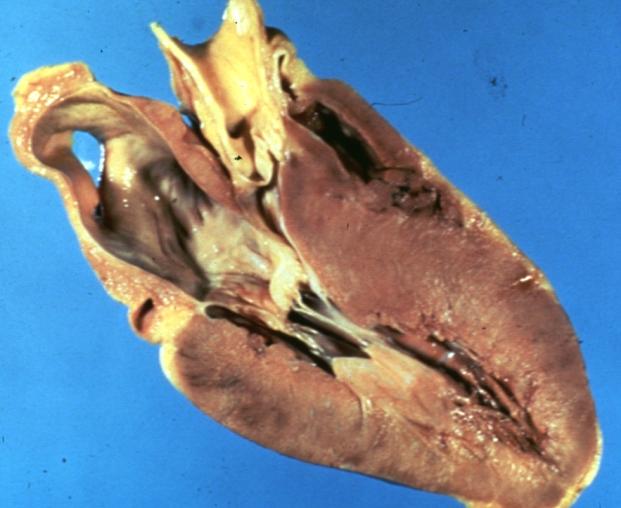 Cardiomyopathy Asymmetrical Septal Hypertrophy
Cardiomyopathy Asymmetrical Septal Hypertrophy


-
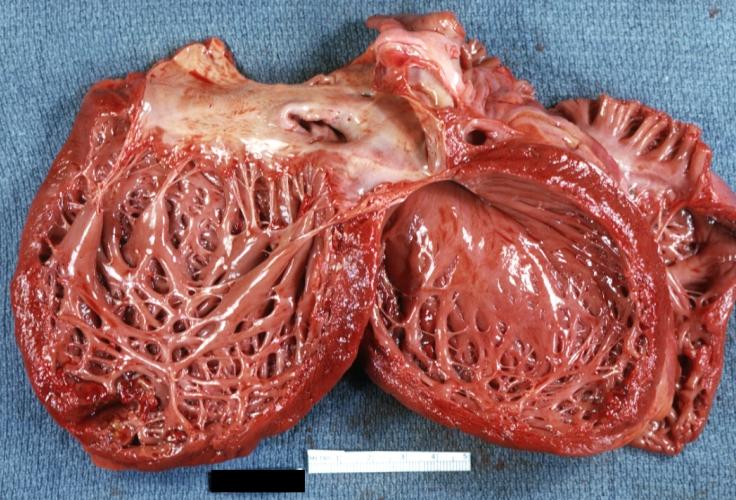 Dilated Cardiomyopathy: Gross opened globular left ventricle natural color (very good example)
Dilated Cardiomyopathy: Gross opened globular left ventricle natural color (very good example) -
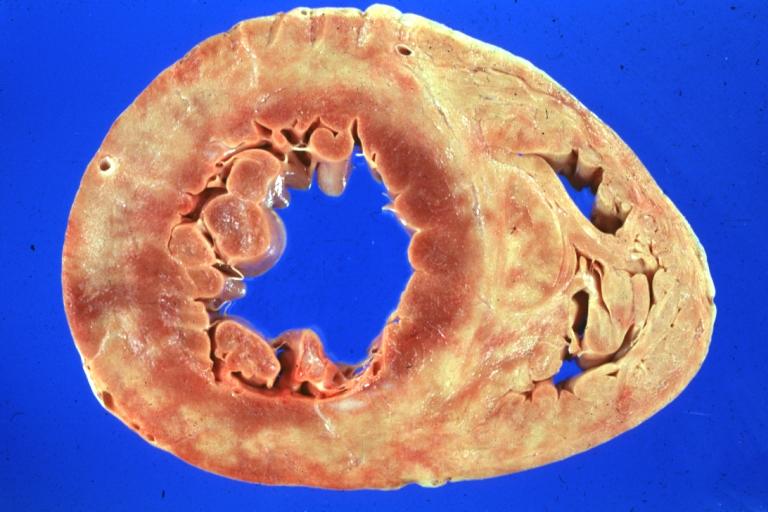 Diabetic Cardiomyopathy: Gross natural color moderately hypertrophied heart shown in horizontal section hyperemic subendocardium has no microscopic lesion long standing type I diabetic patient, no significant coronary artery lesions, congestive heart failure
Diabetic Cardiomyopathy: Gross natural color moderately hypertrophied heart shown in horizontal section hyperemic subendocardium has no microscopic lesion long standing type I diabetic patient, no significant coronary artery lesions, congestive heart failure


-
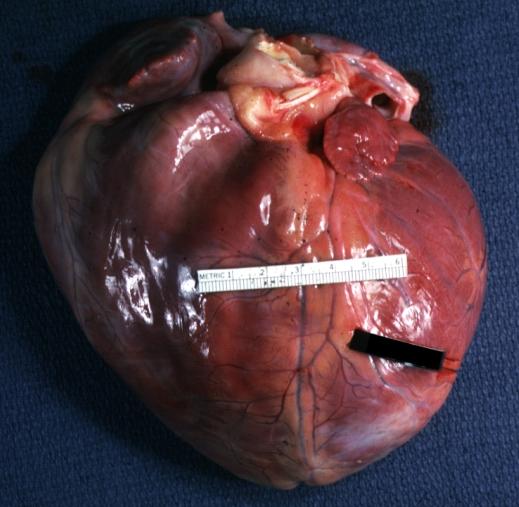 Dilated Cardiomyopathy: Gross natural color external view globular heart 500 gm 24yo female seven pregnancies
Dilated Cardiomyopathy: Gross natural color external view globular heart 500 gm 24yo female seven pregnancies

Microscopic Pathology
[edit | edit source]-
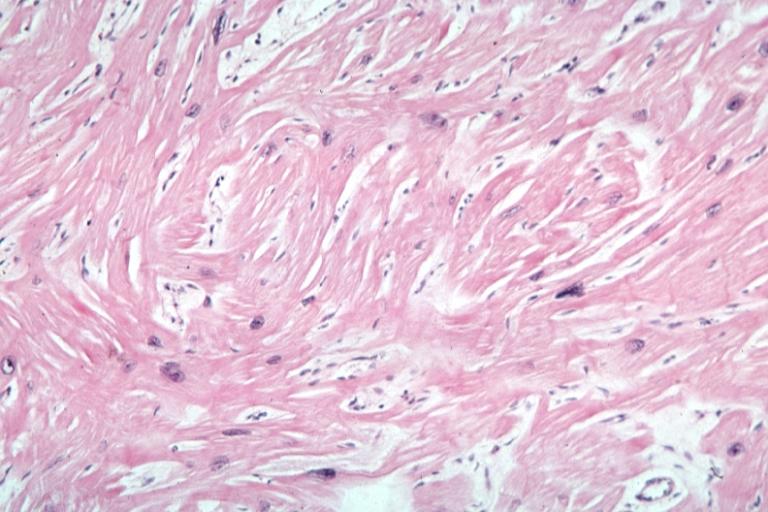 Cardiomyopathy: Micro H&E high mag excellent example myofiber disarray
Cardiomyopathy: Micro H&E high mag excellent example myofiber disarray -
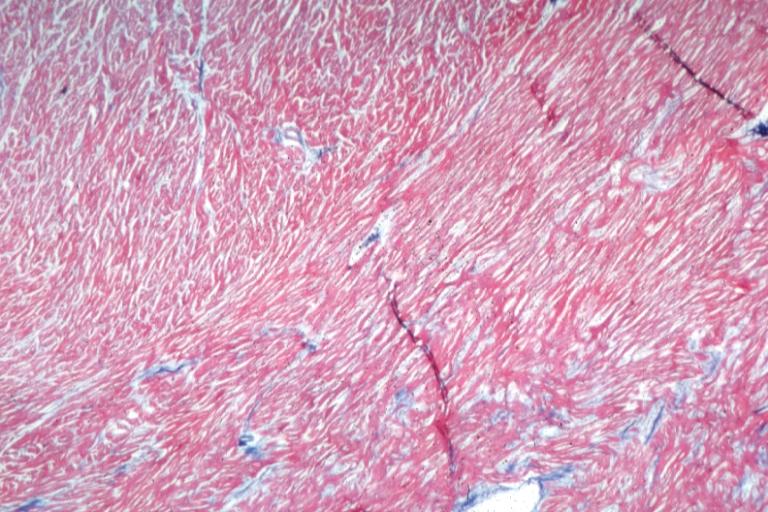 Cardiomyopathy: Micro H&E low mag interventricular septum at junction of normal myofiber orientation with asymmetrical hypertrophy (an excellent example)
Cardiomyopathy: Micro H&E low mag interventricular septum at junction of normal myofiber orientation with asymmetrical hypertrophy (an excellent example)


-
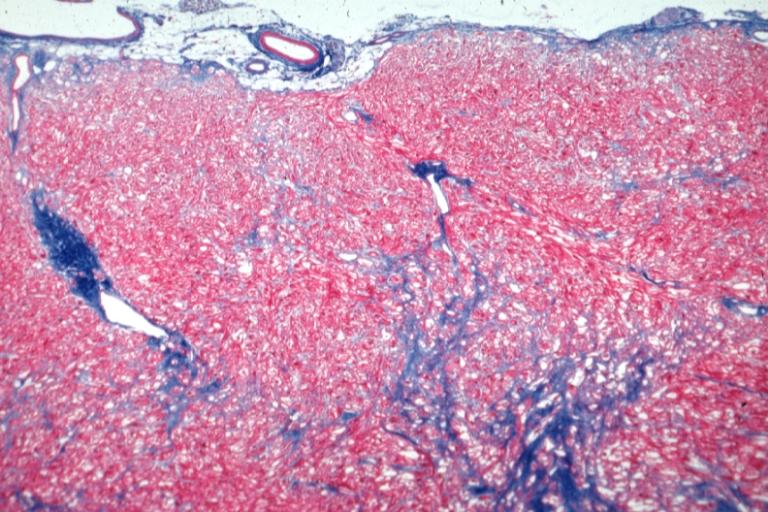 Cardiomyopathy: Micro trichrome low mag bizarre vacuolated fibers with disarray and focal fibrosis excellent low mag epicardial surface
Cardiomyopathy: Micro trichrome low mag bizarre vacuolated fibers with disarray and focal fibrosis excellent low mag epicardial surface -
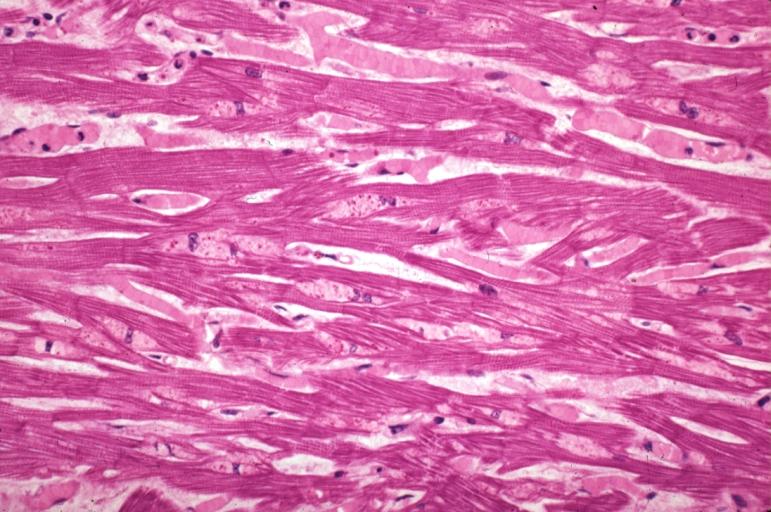 Alcoholic Cardiomyopathy: Micro plastic section lipid in perinuclear area loss of myofibrils
Alcoholic Cardiomyopathy: Micro plastic section lipid in perinuclear area loss of myofibrils


Reference
[edit | edit source]By David Richfield (User:Slashme)When using this image in external works, it may be cited as follows:Richfield, David (2014). "Medical gallery of David Richfield". WikiJournal of Medicine 1 (2). DOI:10.15347/wjm/2014.009. ISSN 2002-4436. - Own work, CC BY-SA 3.0, https://commons.wikimedia.org/w/index.php?curid=2264027
- ↑ 1.0 1.1 1.2 1.3 Invalid
<ref>tag; no text was provided for refs named:0 - ↑ 2.00 2.01 2.02 2.03 2.04 2.05 2.06 2.07 2.08 2.09 2.10 2.11 2.12 2.13 2.14 2.15 2.16 Invalid
<ref>tag; no text was provided for refs named:1 - ↑ 3.00 3.01 3.02 3.03 3.04 3.05 3.06 3.07 3.08 3.09 3.10 3.11 3.12 3.13 3.14 3.15 3.16 3.17 3.18 3.19 3.20 Invalid
<ref>tag; no text was provided for refs named:2 - ↑ 4.00 4.01 4.02 4.03 4.04 4.05 4.06 4.07 4.08 4.09 4.10 4.11 4.12 4.13 4.14 Invalid
<ref>tag; no text was provided for refs named:3 - ↑ Invalid
<ref>tag; no text was provided for refs named:4 - ↑ Invalid
<ref>tag; no text was provided for refs named:5 - ↑ 7.0 7.1 7.2 Invalid
<ref>tag; no text was provided for refs named:6
 EncycloReader
is supported by the
EncycloReader
is supported by the