Chemistry
") From Britannica 11th Edition (1911)
From Britannica 11th Edition (1911) Chemistry (formerly “chymistry”; Gr. χυμεία; for derivation see Alchemy), the natural science which has for its province the study of the composition of substances. In common with physics it includes the determination of properties or characters which serve to distinguish one substance from another, but while the physicist is concerned with properties possessed by all substances and with processes in which the molecules remain intact, the chemist is restricted to those processes in which the molecules undergo some change. For example, the physicist determines the density, elasticity, hardness, electrical and thermal conductivity, thermal expansion, &c.; the chemist, on the other hand, investigates changes in composition, such as may be effected by an electric current, by heat, or when two or more substances are mixed. A further differentiation of the provinces of chemistry and physics is shown by the classifications of matter. To the physicist matter is presented in three leading forms—solids, liquids and gases; and although further subdivisions have been rendered necessary with the growth of knowledge the same principle is retained, namely, a classification based on properties having no relation to composition. The fundamental chemical classification of matter, on the other hand, recognizes two groups of substances, namely, elements, which are substances not admitting of analysis into other substances, and compounds, which do admit of analysis into simpler substances and also of synthesis from simpler substances. Chemistry and physics, however, meet on common ground in a well-defined branch of science, named physical chemistry, which is primarily concerned with the correlation of physical properties and chemical composition, and, more generally, with the elucidation of natural phenomena on the molecular theory.
It may be convenient here to state how the whole subject of chemistry is treated in this edition of the Encyclopaedia Britannica. The present article includes the following sections:—
I. History.—This section is confined to tracing the general trend of the science from its infancy to the foundations of the modern theory. The history of the alchemical period is treated in more detail in the article Alchemy, and of the iatrochemical in the article Medicine. The evolution of the notion of elements is treated under Element; the molecular hypothesis of matter under Molecule; and the genesis of, and deductions from, the atomic theory of Dalton receive detailed analysis in the article Atom.
II. Principles.—This section treats of such subjects as nomenclature, formulae, chemical equations, chemical change and similar subjects. It is intended to provide an introduction, necessarily brief, to the terminology and machinery of the chemist.
III. Inorganic Chemistry.—Here is treated the history of descriptive inorganic chemistry; reference should be made to the articles on the separate elements for an account of their preparation, properties, &c.
IV. Organic Chemistry.—This section includes a brief history of the subject, and proceeds to treat of the principles underlying the structure and interrelations of organic compounds.
V. Analytical Chemistry.—This section treats of the qualitative detection and separation of the metals, and the commoner methods employed in quantitative analysis. The analysis of organic compounds is also noticed.
VI. Physical Chemistry.—This section is restricted to an account of the relations existing between physical properties and chemical composition. Other branches of this subject are treated in the articles Chemical Action; Energetics; Solution; Alloys; Thermochemistry.
I. History
Although chemical actions must have been observed by man in the most remote times, and also utilized in such processes as the extraction of metals from their ores and in the arts of tanning and dyeing, there is no evidence to show that, beyond an unordered accumulation of facts, the early developments of these industries were attended by any real knowledge of the nature of the processes involved. All observations were the result of accident or chance, or possibly in some cases of experimental trial, but there is no record of a theory or even a general classification of the phenomena involved, although there is no doubt that the ancients had a fair knowledge of the properties and uses of the commoner substances. The origin of chemistry is intimately bound up with the arts which we have indicated; in this respect it is essentially an experimental science. A unifying principle of chemical and physical changes was provided by metaphysical conceptions of the structure of matter. We find the notion of “elements,” or primary qualities, which confer upon all species of matter their distinctive qualities by appropriate combination, and also the doctrine that Greek philosophy. matter is composed of minute discrete particles, prevailing in the Greek schools. These “elements,” however, had not the significance of the elements of to-day; they connoted physical appearances or qualities rather than chemical relations; and the atomic theory of the ancients is a speculation based upon metaphysical considerations, having, in its origin, nothing in common with the modern molecular theory, which was based upon experimentally observed properties of gases (see Element; Molecule).
Although such hypotheses could contribute nothing directly to the development of a science which laid especial claim to experimental investigations, yet indirectly they stimulated inquiry into the nature of the “essence” with which the four “elements” were associated. This quinta essentia had been speculated upon by the Greeks, some regarding it as immaterial or aethereal, and others as material; and a school of philosophers termed alchemists arose who attempted the isolation of this essence. The existence of a fundamental principle, unalterable and indestructible, prevailing alike through physical and chemical changes, was generally accepted. Any change which a substance may chance to undergo was simply due to the discarding or taking up of some proportion of the primary “elements” or qualities: of these coverings “water,” “air,” “earth” and “fire” were regarded as clinging most tenaciously to the essence, while “cold,” “heat,” “moistness” and “dryness” were more easily cast aside or assumed. Several origins have been Alchemy. suggested for the word alchemy, and there seems to have been some doubt as to the exact nature and import of the alchemical doctrines. According to M.P.E. Berthelot, “alchemy rested partly on the industrial processes of the ancient Egyptians, partly on the speculative theories of the Greek philosophers, and partly on the mystical reveries of the Gnostics and Alexandrians.” The search for this essence subsequently resolved itself into the desire to effect the transmutation of metals, more especially the base metals, into silver and gold. It seems that this secondary principle became the dominant idea in alchemy, and in this sense the word is used in Byzantine literature of the 4th century; Suidas, writing in the 11th century, defines chemistry as the “preparation of silver and gold” (see Alchemy).
From the Alexandrians the science passed to the Arabs, who made discoveries and improved various methods of separating substances, and afterwards, from the 11th century, became seated in Europe, where the alchemical doctrines were assiduously studied until the 15th and 16th centuries. It is readily understood why men imbued with the authority of tradition should prosecute the search for a substance which would confer unlimited wealth upon the fortunate discoverer. Some alchemists honestly laboured to effect the transmutation and to discover the “philosopher’s stone,” and in many cases believed that they had achieved success, if we may rely upon writings assigned to them. The period, however, is one of literary forgeries; most of the MSS. are of uncertain date and authorship, and moreover are often so vague and mystical that they are of doubtful scientific value, beyond reflecting the tendencies of the age. The retaining of alchemists at various courts shows the high opinion which the doctrines had gained. It is really not extraordinary that Isaac Hollandus was able to indicate the method of the preparation of the “philosopher’s stone” from “adamic” or “virgin” earth, and its action when medicinally employed; that in the writings assigned to Roger Bacon, Raimon Lull, Basil Valentine and others are to be found the exact quantities of it to be used in transmutation; and that George Ripley, in the 15th century, had grounds for regarding its action as similar to that of a ferment.
In the view of some alchemists, the ultimate principles of matter were Aristotle’s four elements; the proximate constituents were a “sulphur” and a “mercury,” the father and mother of the metals; gold was supposed to have attained to the perfection of its nature by passing in succession through the forms of lead, brass and silver; gold and silver were held to contain very pure red sulphur and white quicksilver, whereas in the other metals these materials were coarser and of a different colour. From an analogy instituted between the healthy human being and gold, the most perfect of the metals, silver, mercury, copper, iron, lead and tin, were regarded in the light of lepers that required to be healed.
Notwithstanding the false idea which prompted the researches of the alchemists, many advances were made in descriptive chemistry, the metals and their salts receiving much Iatrochemistry. attention, and several of our important acids being discovered. Towards the 16th century the failure of the alchemists to achieve their cherished purpose, and the general increase of medical knowledge, caused attention to be given to the utilization of chemical preparations as medicines. As early as the 15th century the alchemist Basil Valentine had suggested this application, but the great exponent of this doctrine was Paracelsus, who set up a new definition: “The true use of chemistry is not to make gold but to prepare medicines.” This relation of chemistry to medicine prevailed until the 17th century, and what in the history of chemistry is termed the iatrochemical period (see Medicine) was mainly fruitful in increasing the knowledge of compounds; the contributions to chemical theory are of little value, the most important controversies ranging over the nature of the “elements,” which were generally akin to those of Aristotle, modified so as to be more in accord with current observations. At the same time, however, there were many who, opposed to the Paracelsian definition of chemistry, still laboured at the problem of the alchemists, while others gave much attention to the chemical industries. Metallurgical operations, such as smelting, roasting and refining, were scientifically investigated, and in some degree explained, by Georg Agricola and Carlo Biringuiccio; ceramics was studied by Bernard Palissy, who is also to be remembered as an early worker in agricultural chemistry, having made experiments on the effect of manures on soils and crops; while general technical chemistry was enriched by Johann Rudolf Glauber.1
The second half of the 17th century witnessed remarkable transitions and developments in all branches of natural science, and the facts accumulated by preceding generations Boyle. during their generally unordered researches were replaced by a co-ordination of experiment and deduction. From the mazy and incoherent alchemical and iatrochemical doctrines, the former based on false conceptions of matter, the latter on erroneous views of life processes and physiology, a new science arose—the study of the composition of substances. The formulation of this definition of chemistry was due to Robert Boyle. In his Sceptical Chemist (1662) he freely criticized the prevailing scientific views and methods, with the object of showing that true knowledge could only be gained by the logical application of the principles of experiment and deduction. Boyle’s masterly exposition of this method is his most important contribution to scientific progress. At the same time he clarified the conception of elements and compounds, rejecting the older notions, the four elements of the “vulgar Peripateticks” and the three principles of the “vulgar Stagyrists,” and defining an element as a substance incapable of decomposition, and a compound as composed of two or more elements. He explained chemical combination on the hypotheses that matter consisted of minute corpuscles, that by the coalescence of corpuscles of different substances distinctly new corpuscles of a compound were formed, and that each corpuscle had a certain affinity for other corpuscles.
Although Boyle practised the methods which he expounded, he was unable to gain general acceptance of his doctrine of elements; and, strangely enough, the theory which next dominated chemical thought was an alchemical Phlogistic theory. invention, and lacked the lucidity and perspicuity of Boyle’s views. This theory, named the phlogistic theory, was primarily based upon certain experiments on combustion and calcination, and in effect reduced the number of the alchemical principles, while setting up a new one, a principle of combustibility, named phlogiston (from φλοιστός, burnt). Much discussion had centred about fire or the “igneous principle.” On the one hand, it had been held that when a substance was burned or calcined, it combined with an “air”; on the other hand, the operation was supposed to be attended by the destruction or loss of the igneous principle. Georg Ernst Stahl, following in some measure the views held by Johann Joachim Becher, as, for instance, that all combustibles contain a “sulphur” (which notion is itself of older date than Becher’s terra pinguis), regarded all substances as capable of resolution into two components, the inflammable principle phlogiston, and another element—“water,” “acid” or “earth.” The violence or completeness of combustion was proportional to the amount of phlogiston present. Combustion meant the liberation of phlogiston. Metals on calcination gave calces from which the metals could be recovered by adding phlogiston, and experiment showed that this could generally be effected by the action of coal or carbon, which was therefore regarded as practically pure phlogiston; the other constituent being regarded as an acid. At the hands of Stahl and his school, the phlogistic theory, by exhibiting a fundamental similarity between all processes of combustion and by its remarkable flexibility, came to be a general theory of chemical action. The objections of the antiphlogistonists, such as the fact that calces weigh more than the original metals instead of less as the theory suggests, were answered by postulating that phlogiston was a principle of levity, or even completely ignored as an accident, the change of qualities being regarded as the only matter of importance. It is remarkable that this theory should have gained the esteem of the notable chemists who flourished in the 18th century. Henry Cavendish, a careful and accurate experimenter, was a phlogistonist, as were J. Black, K. W. Scheele, A. S. Marggraf, J. Priestley and many others who might be mentioned.
Descriptive chemistry was now assuming considerable proportions; the experimental inquiries suggested by Boyle were being assiduously developed; and a wealth of observations Lavoisier. was being accumulated, for the explanation of which the resources of the dominant theory were sorely taxed. To quote Antoine Laurent Lavoisier, “... chemists have turned phlogiston into a vague principle, ... which consequently adapts itself to all the explanations for which it may be required. Sometimes this principle has weight, and sometimes it has not; sometimes it is free fire and sometimes it is fire combined with the earthy element; sometimes it passes through the pores of vessels, sometimes these are impervious to it; it explains both causticity and non-causticity, transparency and opacity, colours and their absence; it is a veritable Proteus changing in form at each instant.” Lavoisier may be justly regarded as the founder of modern or quantitative chemistry. First and foremost, he demanded that the balance must be used in all investigations into chemical changes. He established as fundamental that combustion and calcination were attended by an increase of weight, and concluded, as did Jean Rey and John Mayow in the 17th century, that the increase was due to the combination of the metal with the air. The problem could obviously be completely solved only when the composition of the air, and the parts played by its components, had been determined. At all times the air had received attention, especially since van Helmont made his far-reaching investigations on gases. Mayow had suggested the existence of two components, a spiritus nitroaerus which supported combustion, and a spiritus nitri acidi which extinguished fire; J. Priestley and K. W. Scheele, although they isolated oxygen, were fogged by the phlogistic tenets; and H. Cavendish, who had isolated the nitrogen of the atmosphere, had failed to decide conclusively what had really happened to the air which disappeared during combustion.
Lavoisier adequately recognized and acknowledged how much he owed to the researches of others; to himself is due the co-ordination of these researches, and the welding of his results into a doctrine to which the phlogistic theory ultimately succumbed. He burned phosphorus in air standing over mercury, and showed that (1) there was a limit to the amount of phosphorus which could be burned in the confined air, (2) that when no more phosphorus could be burned, one-fifth of the air had disappeared, (3) that the weight of the air lost was nearly equal to the difference in the weights of the white solid produced and the phosphorus burned, (4) that the density of the residual air was less than that of ordinary air. The same results were obtained with lead and tin; and a more elaborate repetition indubitably established their correctness. He also showed that on heating mercury calx alone an “air” was liberated which differed from other “airs,” and was slightly heavier than ordinary air; moreover, the weight of the “air” set free from a given weight of the calx was equal to the weight taken up in forming the calx from mercury, and if the calx be heated with charcoal, the metal was recovered and a gas named “fixed air,” the modern carbon dioxide, was formed. The former experiment had been performed by Scheele and Priestley, who had named the gas “phlogisticated air”; Lavoisier subsequently named it oxygen, regarding it as the “acid producer” (ὀξύς, sour). The theory advocated by Lavoisier came to displace the phlogistic conception; but at first its acceptance was slow. Chemical literature was full of the phlogistic modes of expression—oxygen was “dephlogisticated air,” nitrogen “phlogisticated air,” &c.—and this tended to retard its promotion. Yet really the transition from the one theory to the other was simple, it being only necessary to change the “addition or loss of phlogiston” into the “loss or addition of oxygen.” By his insistence upon the use of the balance as a quantitative check upon the masses involved in all chemical reactions, Lavoisier was enabled to establish by his own investigations and the results achieved by others the principle now known as the “conservation of mass.” Matter can neither be created nor destroyed; however a chemical system be changed, the weights before and after are equal.2 To him is also due a rigorous examination of the nature of elements and compounds; he held the same views that were laid down by Boyle, and with the same prophetic foresight predicted that some of the elements which he himself accepted might be eventually found to be compounds.
It is unnecessary in this place to recapitulate the many results which had accumulated by the end of the 18th century, or to discuss the labours and theories of individual workers since these receive attention under biographical headings; in this article only the salient features in the history of our science can be treated. The beginning of the 19th century was attended by far-reaching discoveries in the nature of the composition of compounds. Investigations proceeded in two directions:—(1) the nature of chemical affinity, (2) the laws Chemical Affinity. of chemical combination. The first question has not yet been solved, although it has been speculated upon from the earliest times. The alchemists explained chemical action by means of such phrases as “like attracts like,” substances being said to combine when one “loved” the other, and the reverse when it “hated” it. Boyle rejected this terminology, which was only strictly applicable to intelligent beings; and he used the word “affinity” as had been previously done by Stahl and others. The modern sense of the word, viz. the force which holds chemically dissimilar substances together (and also similar substances as is seen in di-, tri-, and poly-atomic molecules), was introduced by Hermann Boerhaave, and made more precise by Sir Isaac Newton. The laws of chemical combination were solved, in a measure, by John Dalton, and the solution expressed as Dalton’s “atomic theory.” Lavoisier appears to have assumed that the composition of every chemical compound was constant, and the same opinion was the basis of much experimental inquiry at the hands of Joseph Louis Proust during 1801 to 1809, who vigorously combated the doctrine of Claude Louis Berthollet (Essai de statique chimique, 1803), viz. that fixed proportions of elements and compounds combine only under exceptional conditions, the general rule being that the composition of a compound may vary continuously between certain limits.3
This controversy was unfinished when Dalton published the first part of his New System of Chemical Philosophy in 1808, although the per saltum theory was the most popular. Dalton. Led thereto by speculations on gases, Dalton assumed that matter was composed of atoms, that in the elements the atoms were simple, and in compounds complex, being composed of elementary atoms. Dalton furthermore perceived that the same two elements or substances may combine in different proportions, and showed that these proportions had always a simple ratio to one another. This is the “law of multiple proportions.” He laid down the following arbitrary rules for determining the number of atoms in a compound:—if only one compound of two elements exists, it is a binary compound and its atom is composed of one atom of each element; if two compounds exist one is binary (say A + B) and the other ternary (say A + 2B); if three, then one is binary and the others may be ternary (A + 2B, and 2A + B), and so on. More important is his deduction of equivalent weights, i.e. the relative weights of atoms. He took hydrogen, the lightest substance known, to be the standard. From analyses of water, which he regarded as composed of one atom of hydrogen and one of oxygen, he deduced the relative weight of the oxygen atom to be 6.5; from marsh gas and olefiant gas he deduced carbon = 5, there being one atom of carbon and two of hydrogen in the former and one atom of hydrogen to one of carbon in the latter; nitrogen had an equivalent of 5, and so on.4
The value of Dalton’s generalizations can hardly be overestimated, notwithstanding the fact that in several cases they needed correction. The first step in this direction was effected by the co-ordination of Gay Lussac’s observations on the combining volumes of gases. He discovered that gases always combined in volumes having simple ratios, and that the volume of the product had a simple ratio to the volumes of the reacting gases. For example, one volume of oxygen combined with two of hydrogen to form two volumes of steam, three volumes of hydrogen combined with one of nitrogen to give two volumes of ammonia, one volume of hydrogen combined with one of chlorine to give two volumes of hydrochloric acid. An immediate inference was that the Daltonian “atom” must have parts which enter into combination with parts of other atoms; in other words, there must exist two orders of particles, viz. (1) particles derived by limiting mechanical subdivision, the modern molecule, and (2) particles derived from the first class by chemical subdivision, i.e. particles which are incapable of existing alone, but may exist in combination. Additional evidence as to the structure of the molecule was discussed by Avogadro in 1811, and by Ampere in 1814. From the gas-laws of Boyle and J.A.C. Charles—viz. equal changes in temperature and pressure occasion equal changes in equal volumes of all gases and vapours—Avogadro deduced the law:—Under the same conditions of temperature and pressure, equal volumes of gases contain equal numbers of molecules; and he showed that the relative weights of the molecules are determined as the ratios of the weights of equal volumes, or densities. He established the existence of molecules and atoms as we have defined above, and stated that the number of atoms in the molecule is generally 2, but may be 4, 8, &c. We cannot tell whether his choice of the powers of 2 is accident or design.
Notwithstanding Avogadro’s perspicuous investigation, and a similar exposition of the atom and molecule by A. M. Ampere, the views therein expressed were ignored both by Berzelius. their own and the succeeding generation. In place of the relative molecular weights, attention was concentrated on relative atomic or equivalent weights. This may be due in some measure to the small number of gaseous and easily volatile substances then known, to the attention which the study of the organic compounds received, and especially to the energetic investigations of J. J. Berzelius, who, fired with enthusiasm by the original theory of Dalton and the law of multiple proportions, determined the equivalents of combining ratios of many elements in an enormous number of compounds.5 He prosecuted his labours in this field for thirty years; as proof of his industry it may be mentioned that as early as 1818 he had determined the combining ratios of about two thousand simple and compound substances.
We may here notice the important chemical symbolism or notation
introduced by Berzelius, which greatly contributed to the definite
and convenient representation of chemical composition
Chemical notation.
and the tracing of chemical reactions. The denotation of
elements by symbols had been practised by the alchemists,
and it is interesting to note that the symbols allotted to the well-known
elements are identical with the astrological symbols of the sun and
the other members of the solar system. Gold, the most perfect metal,
had the symbol of the Sun, ☉; silver, the semiperfect metal, had
the symbol of the Moon, ☽; copper, iron and antimony, the
imperfect metals of the gold class, had the symbols of Venus ♀,
Mars ♂, and the Earth ♁; tin and lead, the imperfect metals of
the silver class, had the symbols of Jupiter ♃, and Saturn ♄;
while mercury, the imperfect metal of both the gold and silver
class, had the symbol of the planet, ☿. Torbern Olof Bergman used
an elaborate system in his Opuscula physica et chemica (1783); the
elements received symbols composed of circles, arcs of circles, and
lines, while certain class symbols, such as  for metals,
for metals,  for acids,
for acids,
 for alkalies,
for alkalies,  for salts,
for salts, 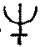 for calces, &c., were used. Compounds
were represented by copulating simpler symbols, e.g. mercury calx
was
for calces, &c., were used. Compounds
were represented by copulating simpler symbols, e.g. mercury calx
was 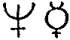 .6 Bergman’s symbolism was obviously cumbrous, and
the system used in 1782 by Lavoisier was equally abstruse, since the
forms gave no clue as to composition; for instance water, oxygen,
and nitric acid were
.6 Bergman’s symbolism was obviously cumbrous, and
the system used in 1782 by Lavoisier was equally abstruse, since the
forms gave no clue as to composition; for instance water, oxygen,
and nitric acid were 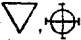 , and
, and 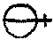 .
.
A partial clarification was suggested in 1787 by J.H. Hassenfratz and Adet, who assigned to each element a symbol, and to each compound a sign which should record the elements present and their relative quantities. Straight lines and semicircles were utilized for the non-metallic elements, carbon, nitrogen, phosphorus and sulphur (the “simple acidifiable bases” of Lavoisier), and circles enclosing the initial letters of their names for the metals. The “compound acidifiable bases,” i.e. the hypothetical radicals of acids, were denoted by squares enclosing the initial letter of the base; an alkali was denoted by a triangle, and the particular alkali by inserting the initial letter. Compounds were denoted by joining the symbols of the components, and by varying the manner of joining compounds of the same elements were distinguished. The symbol \/ was used to denote a liquid, and a vertical line to denote a gas. As an example of the complexity of this system we may note the five oxides of nitrogen, which were symbolized as
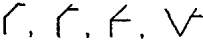 and
and 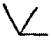 ,
,
the first three representing the gaseous oxides, and the last two the liquid oxides.
A great advance was made by Dalton, who, besides introducing simpler symbols, regarded the symbol as representing not only the element or compound but also one atom of that element or compound; in other words, his symbol denoted equivalent weights.7 This system, which permitted the correct representation of molecular composition, was adopted by Berzelius in 1814, who, having replaced the geometric signs of Dalton by the initial letter (or letters) of the Latin names of the elements, represented a compound by placing a plus sign between the symbols of its components, and the number of atoms of each component (except in the case of only one atom) by placing Arabic numerals before the symbols; for example, copper oxide was Cu+O, sulphur trioxide S+3O. If two compounds combined, the + signs of the free compounds were discarded, and the number of atoms denoted by an Arabic index placed after the elements, and from these modified symbols the symbol of the new compound was derived in the same manner as simple compounds were built up from their elements. Thus copper sulphate was CuO + SO3, potassium sulphate 2SO3 + PoO2 (the symbol Po for potassium was subsequently discarded in favour of K from kalium). At a later date Berzelius denoted an oxide by dots, equal in number to the number of oxygen atoms present, placed over the element; this notation survived longest in mineralogy. He also introduced barred symbols, i.e. letters traversed by a horizontal bar, to denote the double atom (or molecule). Although the system of Berzelius has been modified and extended, its principles survive in the modern notation.
In the development of the atomic theory and the deduction of the atomic weights of elements and the formulae of compounds, Dalton’s arbitrary rules failed to find complete acceptance. Extension of the atomic theory. Berzelius objected to the hypothesis that if two elements form only one compound, then the atoms combine one and one; and although he agreed with the adoption of simple rules as a first attempt at representing a compound, he availed himself of other data in order to gain further information as to the structure of compounds. For example, at first he represented ferrous and ferric oxides by the formulae FeO2, FeO3, and by the analogy of zinc and other basic oxides he regarded these substances as constituted similarly to FeO2, and the acidic oxides alumina and chromium oxide as similar to FeO3. He found, however, that chromic acid, which he had represented as CrO6, neutralized a base containing 1/3 the quantity of oxygen. He inferred that chromic acid must contain only three atoms of oxygen, as did sulphuric acid SO3; consequently chromic oxide, which contains half the amount of oxygen, must be Cr2O3, and hence ferric oxide must be Fe2O3. The basic oxides must have the general formula MO. To these results he was aided by the law of isomorphism formulated by E. Mitscherlich in 1820; and he confirmed his conclusions by showing the agreement with the law of atomic heat formulated by Dulong and Petit in 1819.
While successfully investigating the solid elements and their compounds gravimetrically, Berzelius was guilty of several inconsistencies in his views on gases. He denied that gaseous atoms could have parts, although compound gases could. This attitude was due to his adherence to the “dualistic theory” of the structure of substances, which he deduced from electrochemical researches. From the behaviour of substances on electrolysis (q.v.) he assumed that all substances had two components, one bearing a negative charge, the other a positive charge. Combination was associated with the coalescence of these charges, and the nature of the resulting compound showed the nature of the residual electricity. For example, positive iron combined with negative oxygen to form positive ferrous oxide; positive sulphur combined with negative oxygen to form negative sulphuric acid; positive ferrous oxide combined with negative sulphuric acid to form neutral ferrous sulphate. Berzelius elevated this theory to an important position in the history of our science. He recognized that if an elementary atom had parts, his theory demanded that these parts should carry different electric charges when they entered into reaction, and the products of the reaction should vary according as a positive or negative atom entered into combination. For instance if the reaction 2H2 + O2 = H2O + H2O be true, the molecules of water should be different, for a negative oxygen atom would combine in one case, and a positive oxygen atom in the other. Hence the gaseous atoms of hydrogen and oxygen could not have parts. A second inconsistency was presented when he was compelled by the researches of Dumas to admit Avogadro’s hypothesis; but here he would only accept it for the elementary gases, and denied it for other substances. It is to be noticed that J.B. Dumas did not adopt the best methods for emphasizing his discoveries. His terminology was vague and provoked caustic criticism from Berzelius; he assumed that all molecules contained two atoms, and consequently the atomic weights deduced from vapour density determinations of sulphur, mercury, arsenic, and phosphorus were quite different from those established by gravimetric and other methods.
Chemists gradually tired of the notion of atomic weights on account of the uncertainty which surrounded them; and the suggestion made by W.H. Wollaston as early as 1814 to deal only with “equivalents,” i.e. the amount of an element which can combine with or replace unit weight of hydrogen, came into favour, being adopted by L. Gmelin in his famous text-book.
Simultaneously with this discussion of the atom and molecule, great controversy was ranging over the constitution of compounds, more particularly over the carbon or organic Atomic and molecular weights. compounds. This subject is discussed in section IV., Organic Chemistry.The gradual accumulation of data referring to organic compounds brought in its train a revival of the discussion of atoms and molecules. A. Laurent and C.F. Gerhardt attempted a solution by investigating chemical reactions. They assumed the atom to be the smallest part of matter which can exist in combination, and the molecule to be the smallest part which can enter into a chemical reaction. Gerhardt found that reactions could be best followed if one assumed the molecular weight of an element or compound to be that weight which occupied the same volume as two unit weights of hydrogen, and this assumption led him to double the equivalents accepted by Gmelin, making H = 1, O = 16, and C = 12, thereby agreeing with Berzelius, and also to halve the values given by Berzelius to many metals. Laurent generally agreed, except when the theory compelled the adoption of formulae containing fractions of atoms; in such cases he regarded the molecular weight as the weight occupying a volume equal to four unit weights of hydrogen. The bases upon which Gerhardt and Laurent founded their views were not sufficiently well grounded to lead to the acceptance of their results; Gerhardt himself returned to Gmelin’s equivalents in his Lehrbuch der Chemie (1853) as they were in such general use.
In 1860 there prevailed such a confusion of hypotheses as to the atom and molecule that a conference was held at Karlsruhe to discuss the situation. At the conclusion of the sitting, Lothar Meyer obtained a paper written by Stanislas Cannizzaro in 1858 wherein was found the final link required for the determination of atomic weights. This link was the full extension of Avogadro’s theory to all substances, Cannizzaro showing that chemical reactions in themselves would not suffice. He chose as his unit of reference the weight of an atom of hydrogen, i.e. the weight contained in a molecule of hydrochloric acid, thus differing from Avogadro who chose the weight of a hydrogen molecule. From a study of the free elements Cannizzaro showed that an element may have more than one molecular weight; for example, the molecular weight of sulphur varied with the temperature. And from the study of compounds he showed that each element occurred in a definite weight or in some multiple of this weight. He called this proportion the “atom,” since it invariably enters compounds without division, and the weight of this atom is the atomic weight. This generalization was of great value inasmuch as it permitted the deduction of the atomic weight of a non-gasifiable element from a study of the densities of its gasifiable compounds.
From the results obtained by Laurent and Gerhardt and their predecessors it immediately followed that, while an element could have but one atomic weight, it could have several equivalent weights. From a detailed study of organic compounds Gerhardt had promulgated a “theory of types” which represented a fusion of the older radical and type theories. This theory brought together, as it were, the most varied compounds, and stimulated inquiry into many fields. According to this theory, an element in a compound had a definite saturation capacity, an idea very old in itself, being framed in the law of multiple Valency. proportions. These saturation capacities were assiduously studied by Sir Edward Frankland, who from the investigation, not of simple inorganic compounds, but of the organo-metallic derivatives, determined the kernel of the theory of valency. Frankland showed that any particular element preferentially combined with a definite number (which might vary between certain limits) of other atoms; for example, some atoms always combined with one atom of oxygen, some with two, while with others two atoms entered into combination with one of oxygen. If an element or radical combined with one atom of hydrogen, it was termed monovalent; if with two (or with one atom of oxygen, which is equivalent to two atoms of hydrogen) it was divalent, and so on. The same views were expressed by Cannizzaro, and also by A.W. von Hofmann, who materially helped the acceptance of the doctrine by the lucid exposition in his Introduction to Modern Chemistry, 1865.
The recognition of the quadrivalency of carbon by A. Kekulé was the forerunner of his celebrated benzene theory in particular, and of the universal application of structural formulae to the representation of the most complex organic compounds equally lucidly as the representation of the simplest salts. Alexander Butlerow named the “structure theory,” and contributed much to the development of the subject. He defined structure “as the manner of the mutual linking of the atoms in the molecule,” but denied that any such structure could give information as to the orientation of the atoms in space. He regarded the chemical properties of a substance as due to (1) the chemical atoms composing it, and (2) the structure, and he asserted that while different compounds might have the same components (isomerism), yet only one compound could have a particular structure. Identity in properties necessitated identity in structure.
While the principle of varying valency laid down by Frankland is still retained, Butlerow’s view that structure had no spatial significance has been modified. The researches of L. Pasteur, J.A. Le Bel, J. Wislicenus, van’t Hoff and others showed that substances having the same graphic formulae vary in properties and reactions, and consequently the formulae need modification in order to exhibit these differences. Such isomerism, named stereoisomerism (q.v.), has been assiduously developed during recent years; it prevails among many different classes of organic compounds and many examples have been found in inorganic chemistry.
The theory of valency as a means of showing similarity of properties and relative composition became a dominant feature of chemical theory, the older hypotheses of types, radicals, &c. being more or less discarded. We have seen how its Periodic law. utilization in the “structure theory” permitted great clarification, and attempts were not wanting for the deduction of analogies or a periodicity between elements. Frankland had recognized the analogies existing between the chemical properties of nitrogen, phosphorus, arsenic and antimony, noting that they act as tri- or penta-valent. Carbon was joined with silicon, zirconium and titanium, while boron, being trivalent, was relegated to another group. A general classification of elements, however, was not realized by Frankland, nor even by Odling, who had also investigated the question from the valency standpoint. The solution came about by arranging the elements in the order of their atomic weights, tempering the arrangement with the results deduced from the theory of valencies and experimental observations. Many chemists contributed to the establishment of such a periodicity, the greatest advances being made by John Newlands in England, Lothar Meyer in Germany, and D.J. Mendeléeff in St Petersburg. For the development of this classification see Element.
In the above sketch we have briefly treated the history of the main tendencies of our science from the earliest times to the establishment of the modern laws and principles. We Summary. have seen that the science took its origin in the arts practised by the Egyptians, and, having come under the influence of philosophers, it chose for its purpose the isolation of the quinta essentia, and subsequently the “art of making gold and silver.” This spirit gave way to the physicians, who regarded “chemistry as the art of preparing medicines,” a denotation which in turn succumbed to the arguments of Boyle, who regarded it as the “science of the composition of substances,” a definition which adequately fits the science to-day. We have seen how his classification of substances into elements and compounds, and the definitions which he assigned to these species, have similarly been retained; and how Lavoisier established the law of the “conservation of mass,” overthrew the prevailing phlogistic theory, and became the founder of modern chemistry by the overwhelming importance which he gave to the use of the balance. The development of the atomic theory and its concomitants—the laws of chemical combination and the notion of atoms and equivalents—at the hands of Dalton and Berzelius, the extension to the modern theory of the atom and molecule, and to atomic and molecular weights by Avogadro, Ampère, Dumas, Laurent, Gerhardt, Cannizzaro and others, have been noted. The structure of the molecule, which mainly followed investigations in organic compounds, Frankland’s conception of valency, and finally the periodic law, have also been shown in their chronological order. The principles outlined above constitute the foundations of our science; and although it may happen that experiments may be made with which they appear to be not in complete agreement, yet in general they constitute a body of working hypotheses of inestimable value.
Chemical Education.—It is remarkable that systematic instruction in the theory and practice of chemistry only received earnest attention in our academic institutions during the opening decades of the 19th century. Although for a long time lecturers and professors had been attached to universities, generally their duties had also included the study of physics, mineralogy and other subjects, with the result that chemistry received scanty encouragement. Of practical instruction there was none other than that to be gained in a few private laboratories and in the shops of apothecaries. The necessity for experimental demonstration and practical instruction, in addition to academic lectures, appears to have been urged by the French chemists L.N. Vauquelin, Gay Lussac, Thénard, and more especially by A.F. Fourcroy and G.F. Rouelle, while in England Humphry Davy expounded the same idea in the experimental demonstrations which gave his lectures their brilliant charm. But the real founder of systematic instruction in our science was Justus von Liebig, who, having accepted the professorship at Giessen in 1824, made his chemical laboratory and course of instruction the model of all others. He emphasized that the practical training should include (1) the qualitative and quantitative analysis of mixtures, (2) the preparation of substances according to established methods, (3) original research—a course which has been generally adopted. The pattern set by Liebig at Giessen was adopted by F. Wöhler at Göttingen in 1836, by R.W. Bunsen at Marburg in 1840, and by O.L. Erdmann at Leipzig in 1843; and during the ’fifties and ’sixties many other laboratories were founded. A new era followed the erection of the laboratories at Bonn and Berlin according to the plans of A.W. von Hofmann in 1867, and of that at Leipzig, designed by Kolbe in 1868. We may also mention the famous laboratory at Munich designed by A. von Baeyer in 1875.
In Great Britain the first public laboratory appears to have been opened in 1817 by Thomas Thomson at Glasgow. But the first important step in providing means whereby students could systematically study chemistry was the foundation of the College of Chemistry in 1845. This institution was taken over by the Government in 1853, becoming the Royal College of Chemistry, and incorporated with the Royal School of Mines; in 1881 the names were changed to the Normal School of Science and Royal School of Mines, and again in 1890 to the Royal College of Science. In 1907 it was incorporated in the Imperial College of Science and Technology. Under A.W. von Hofmann, who designed the laboratories and accepted the professorship in 1845 at the instigation of Prince Albert, and under his successor (in 1864) Sir Edward Frankland, this institution became one of the most important centres of chemical instruction. Oxford and Cambridge sadly neglected the erection of convenient laboratories for many years, and consequently we find technical schools and other universities having a far better equipment and offering greater facilities. In the provinces Victoria University at Manchester exercised the greater impetus, numbering among its professors Sir W.H. Perkin and Sir Henry Roscoe.
In America public laboratory instruction was first instituted at Yale College during the professorship of Benjamin Silliman. To the great progress made in recent years F.W. Clarke, W. Gibbs, E.W. Morley, Ira Remsen, and T.W. Richards have especially contributed.
In France the subject was almost entirely neglected until late in the 19th century. The few laboratories existing in the opening decades were ill-fitted, and the exorbitant fees constituted a serious bar to general instruction, for these institutions received little government support. In 1869 A. Wurtz reported the existence of only one efficient laboratory in France, namely the École Normale Supérieure, under the direction of H. Sainte Claire Deville. During recent years chemistry has become one of the most important subjects in the curriculum of technical schools and universities, and at the present time no general educational institution is complete until it has its full equipment of laboratories and lecture theatres.
Chemical Literature.—The growth of chemical literature since the publication of Lavoisier’s famous Traité de chimie in 1789, and of Berzelius’ Lehrbuch der Chemie in 1808-1818, has been enormous. These two works, and especially the latter, were the models followed by Thénard, Liebig, Strecker, Wöhler and many others, including Thomas Graham, upon whose Elements of Chemistry was founded Otto’s famous Lehrbuch der Chemie, to which H. Kopp contributed the general theoretical part, Kolbe the organic, and Buff and Zamminer the physico-chemical. Organic chemistry was especially developed by the publication of Gerhardt’s Traité de chimie organique in 1853-1856, and of Kekulé’s Lehrbuch der organischen Chemie in 1861-1882. General theoretical and physical chemistry was treated with conspicuous acumen by Lothar Meyer in his Moderne Theorien, by W. Ostwald in his Lehrbuch der allgem. Chemie (1884-1887), and by Nernst in his Theoretische Chemie. In English, Roscoe and Schorlemmer’s Treatise on Chemistry is a standard work; it records a successful attempt to state the theories and facts of chemistry, not in condensed epitomes, but in an easily read form. The Traité de chimie minérale, edited by H. Moissan, and the Handbuch der anorganischen Chemie, edited by Abegg, are of the same type. O. Dammer’s Handbuch der anorganischen Chemie and F. Beilstein’s Handbuch der organischen Chemie are invaluable works of reference. Of the earlier encyclopaedias we may notice the famous Handwörterbuch der reinen und angewandten Chemie, edited by Liebig; Frémy’s Encyclopédie de chimie, Wurtz’s Dictionnaire de chimie pure et appliquée, Watts’ Dictionary of Chemistry, and Ladenburg’s Handwörterbuch der Chemie.
The number of periodicals devoted to chemistry has steadily increased since the early part of the 19th century. In England the most important is the Journal of the Chemical Society of London, first published in 1848. Since 1871 abstracts of papers appearing in the other journals have been printed. In 1904 a new departure was made in issuing Annual Reports, containing résumés of the most important researches of the year. The Chemical News, founded by Sir W. Crookes in 1860, may also be noted. In America the chief periodical is the American Chemical Journal, founded in 1879. Germany is provided with a great number of magazines. The Berichte der deutschen chemischen Gesellschaft, published by the Berlin Chemical Society, the Chemisches Centralblatt, which is confined to abstracts of papers appearing in other journals, the Zeitschrift für Chemie, and Liebig’s Annalen der Chemie are the most important of the general magazines. Others devoted to special phases are the Journal für praktische Chemie, founded by Erdmann in 1834, the Zeitschrift für anorganische Chemie and the Zeitschrift für physikalische Chemie. Mention may also be made of the invaluable Jahresberichte and the Jahrbuch der Chemie. In France, the most important journals are the Annales de chimie et de physique, founded in 1789 with the title Annales de chimie, and the Comptes rendus, published weekly by the Académie française since 1835.
II. General Principles
The substances with which the chemist has to deal admit of classification into elements and compounds. Of the former about eighty may be regarded as well characterized, although many more have been described.
Elements.—The following table gives the names, symbols and atomic weights of the perfectly characterized elements:—
International Atomic Weights, 1910.
| Name. | Symbol. | Atomic Weights. O=16. | Name. | Symbol. | Atomic Weights. O=16. | |||
| Aluminium | Al | 27. | 1 | Mercury | Hg | 200. | 0 | |
| Antimony | Sb | 120. | 2 | Molybdenum | Mo | 96. | 0 | |
| Argon | A | 39. | 9 | Neodymium | Nd | 144. | 3 | |
| Arsenic | As | 74. | 96 | Neon | Ne | 20 | ||
| Barium | Ba | 137. | 37 | Nickel | Ni | 58. | 68 | |
| Beryllium or | Be } | 9. | 1 | Nitrogen | N | 14. | 01 | |
| Glucinum | Gl } | Osmium | Os | 190. | 9 | |||
| Bismuth | Bi | 208. | 0 | Oxygen | O | 16. | 00 | |
| Boron | B | 11. | 0 | Palladium | Pd | 106. | 7 | |
| Bromine | Br | 79. | 92 | Phosphorus | P | 31. | 0 | |
| Cadmium | Cd | 112. | 40 | Platinum | Pt | 195. | 0 | |
| Caesium | Cs | 132. | 81 | Potassium | K | 39. | 10 | |
| Calcium | Ca | 40. | 09 | Praseodymium | Pr | 140. | 6 | |
| Carbon | C | 12. | 0 | Radium | Ra | 226. | 4 | |
| Cerium | Ce | 140. | 25 | Rhodium | Rh | 102. | 9 | |
| Chlorine | Cl | 35. | 46 | Rubidium | Rb | 85. | 45 | |
| Chromium | Cr | 52. | 0 | Ruthenium | Ru | 101. | 7 | |
| Cobalt | Co | 58. | 97 | Samarium | Sa | 150. | 4 | |
| Columbium | Cb } | 93. | 5 | Scandium | Sc | 44. | 1 | |
| or Niobium | Nb } | Selenium | Se | 79. | 2 | |||
| Copper | Cu | 63. | 57 | Silicon | Si | 28. | 3 | |
| Dysprosium | Dy | 162. | 5 | Silver | Ag | 107. | 88 | |
| Erbium | Er | 167. | 4 | Sodium | Na | 23. | 0 | |
| Europium | Eu | 152. | 0 | Strontium | Sr | 87. | 62 | |
| Fluorine | F | 19. | 0 | Sulphur | S | 32. | 07 | |
| Gadolinium | Gd | 157. | 3 | Tantalum | Ta | 181. | 0 | |
| Gallium | Ga | 69. | 9 | Tellurium | Te | 127. | 5 | |
| Germanium | Ge | 72. | 5 | Terbium | Tb | 159. | 2 | |
| Gold | Au | 197. | 2 | Thallium | Tl | 204. | 0 | |
| Helium | He | 4. | 0 | Thorium | Th | 232. | 42 | |
| Hydrogen | H | 1. | 008 | Thulium | Tm | 168. | 5 | |
| Indium | In | 114. | 8 | Tin | Sn | 119. | 0 | |
| Iodine | I | 126. | 92 | Titanium | Ti | 48. | 1 | |
| Iridium | Ir | 193. | 1 | Tungsten | W | 184. | 0 | |
| Iron | Fe | 55. | 85 | Uranium | U | 238. | 5 | |
| Krypton | Kr | 83. | 0 | Vanadium | V | 51. | 2 | |
| Lanthanum | La | 139. | 0 | Xenon | Xe | 130. | 7 | |
| Lead | Pb | 207. | 10 | Ytterbium | ||||
| Lithium | Li | 7. | 00 | (Neoytterbium) | Yb | 172 | ||
| Lutecium | Lu | 174 | Yttrium | Y | 89. | 0 | ||
| Magnesium | Mg | 24. | 32 | Zinc | Zn | 65. | 37 | |
| Manganese | Mn | 54. | 93 | Zirconium | Zr | 90. | 6 | |
The elements are usually divided into two classes, the metallic and the non-metallic elements; the following are classed as non-metals, and the remainder as metals:—
| Hydrogen | Oxygen | Boron | Neon |
| Chlorine | Sulphur | Carbon | Krypton |
| Bromine | Selenium | Silicon | Xenon |
| Iodine | Tellurium | Phosphorus | elium |
| Fluorine | Nitrogen | Argon |
Of these hydrogen, chlorine, fluorine, oxygen, nitrogen, argon, neon, krypton, xenon and helium are gases, bromine is a liquid, and the remainder are solids. All the metals are solids at ordinary temperatures with the exception of mercury, which is liquid. The metals are mostly bodies of high specific gravity; they exhibit, when polished, a peculiar brilliancy or metallic lustre, and they are good conductors of heat and electricity; the non-metals, on the other hand, are mostly bodies of low specific gravity, and bad conductors of heat and electricity, and do not exhibit metallic lustre. The non-metallic elements are also sometimes termed metalloids, but this appellation, which signifies metal-like substances (Gr. εἰδος, like), strictly belongs to certain elements which do not possess the properties of the true metals, although they more closely resemble them than the non-metals in many respects; thus, selenium and tellurium, which are closely allied to sulphur in their chemical properties, although bad conductors of heat and electricity, exhibit metallic lustre and have relatively high specific gravities. But when the properties of the elements are carefully contrasted together it is found that no strict line of demarcation can be drawn dividing them into two classes; and if they are arranged in a series, those which are most closely allied in properties being placed next to each other, it is observed that there is a more or less regular alteration in properties from term to term in the series.
When binary compounds, or compounds of two elements, are decomposed by an electric current, the two elements make their appearance at opposite poles. Those elements which are disengaged at the negative pole are termed electro-positive, or positive, or basylous elements, whilst those disengaged at the positive pole are termed electro-negative, or negative, or chlorous elements. But the difference between these two classes of elements is one of degree only, and they gradually merge into each other; moreover the electric relations of elements are not absolute, but vary according to the state of combination in which they exist, so that it is just as impossible to divide the elements into two classes according to this property as it is to separate them into two distinct classes of metals and non-metals. The following, however, are negative towards the remaining elements which are more or less positive:—Fluorine, chlorine, bromine, iodine, oxygen, sulphur, selenium, tellurium.
The metals may be arranged in a series according to their power of displacing one another in salt solutions, thus Cs, Rb, K, Na, Mg, Al, Mn, Zn, Cd, Tl, Fe, Co, Ni, Sn, Pb, (H), Sb, Bi, As, Cu, Hg, Ag, Pd, Pt, Au.
Elements which readily enter into reaction with each other, and which develop a large amount of heat on combination, are said to have a powerful affinity for each other. The tendency of positive elements to unite with positive elements, or of negative elements to unite with negative elements, is much less than that of positive elements to unite with negative elements, and the greater the difference in properties between two elements the more powerful is their affinity for each other. Thus, the affinity of hydrogen and oxygen for each other is extremely powerful, much heat being developed by the combination of these two elements; when binary compounds of oxygen are decomposed by the electric current, the oxygen invariably appears at the positive pole, being negative to all other elements, but the hydrogen of hydrogen compounds is always disengaged at the negative pole. Hydrogen and oxygen are, therefore, of very opposite natures, and this is well illustrated by the circumstance that oxygen combines, with very few exceptions, with all the remaining elements, whilst compounds of only a limited number with hydrogen have been obtained.
Compounds.—A chemical compound contains two or more elements; consequently it should be possible to analyse it, i.e. separate it into its components, or to synthesize it, i.e. build it up from its components. In general, a compound has properties markedly different from those of the elements of which it is composed.
Laws of Chemical Combination.—A molecule may be defined as the smallest part of a substance which can exist alone; an atom as the smallest part of a substance which can exist in combination. The molecule of every compound must obviously contain at least two atoms, and generally the molecules of the elements are also polyatomic, the elements with monatomic molecules (at moderate temperatures) being mercury and the gases of the argon group. The laws of chemical combination are as follows:—
1. Law of Definite Proportions.—The same compound always contains the same elements combined together in the same mass proportion. Silver chloride, for example, in whatever manner it may be prepared, invariably consists of chlorine and silver in the proportions by weight of 35.45 parts of the former and 107.93 of the latter.
2. Law of Multiple Proportions.—When the same two elements combine together to form more than one compound, the different masses of one of the elements which unite with a constant mass of the other, bear a simple ratio to one another. Thus, 1 part by weight of hydrogen unites with 8 parts by weight of oxygen, forming water, and with 16 or 8 × 2 parts of oxygen, forming hydrogen peroxide. Again, in nitrous oxide we have a compound of 8 parts by weight of oxygen and 14 of nitrogen; in nitric oxide a compound of 16 or 8 × 2 parts of oxygen and 14 of nitrogen; in nitrous anhydride a compound of 24 or 8 × 3 parts of oxygen and 14 of nitrogen; in nitric peroxide a compound of 32 or 8 × 4 parts of oxygen and 14 of nitrogen; and lastly, in nitric anhydride a compound of 40 or 8 × 5 parts of oxygen and 14 of nitrogen.
3. Law of Reciprocal Proportions.—The masses of different elements which combine separately with one and the same mass of another element, are either the same as, or simple multiples of, the masses of these different elements which combine with each other. For instance, 35.45 parts of chlorine and 79.96 parts of bromine combine with 107.93 parts of silver; and when chlorine and bromine unite it is in the proportion of 35.45 parts of the former to 79.96 parts of the latter. Iodine unites with silver in the proportion of 126.97 parts to 107.93 parts of the latter, but it combines with chlorine in two proportions, viz. in the proportion of 126.97 parts either to 35.45 or to three times 35.45 parts of chlorine.
There is a fourth law of chemical combination which only applies to gases. This law states that:—gases combine with one another in simple proportions by volume, and the volume of the product (if gaseous) has a simple ratio to the volumes of the original mixtures; in other words, the densities of gases are simply related to their combining weights.
Nomenclature.—If a compound contains two atoms it is termed a binary compound, if three a ternary, if four a quaternary, and so on. Its systematic name is formed by replacing the last syllable of the electro-negative element by ide and prefixing the name of the other element. For example, compounds of oxygen are oxides, of chlorine, chlorides, and so on. If more than one compound be formed from the same two elements, the difference is shown by prefixing such words as mono-, di-, tri-, sesqui-, per-, sub-, &c., to the last part of the name, or the suffixes -ous and -ic may be appended to the name of the first element. For example take the oxides of nitrogen, N2O, NO, N2O3, NO2, N2O5; these are known respectively as nitrous oxide, nitric oxide, nitrogen trioxide, nitrogen peroxide and nitrogen pentoxide. The affixes -ous and sub- refer to the compounds containing more of the positive element, -ic and per- to those containing less.
An acid (q.v.) is a compound of hydrogen, which element can be replaced by metals, the hydrogen being liberated, giving substances named salts. An alkali or base is a substance which neutralizes an acid with the production of salts but with no evolution of hydrogen. A base may be regarded as water in which part of the hydrogen is replaced by a metal, or by a radical which behaves as a metal. (The term radical is given to a group of atoms which persist in chemical changes, behaving as if the group were an element; the commonest is the ammonium group, NH4, which forms salts similar to the salts of sodium and potassium.) If the acid contains no oxygen it is a hydracid, and its systematic name is formed from the prefix hydro- and the name of the other element or radical, the last syllable of which has been replaced by the termination -ic. For example, the acid formed by hydrogen and chlorine is termed hydrochloric acid (and sometimes hydrogen chloride). If an acid contains oxygen it is termed an oxyacid. The nomenclature of acids follows the same general lines as that for binary compounds. If one acid be known its name is formed by the termination -ic, e.g. carbonic acid; if two, the one containing the less amount of oxygen takes the termination -ous and the other the termination -ic, e.g. nitrous acid, HNO2, nitric acid, HNO3. If more than two be known, the one inferior in oxygen content has the prefix hypo- and the termination -ous, and the one superior in oxygen content has the prefix per- and the termination -ic. This is illustrated in the four oxyacids of chlorine, HClO, HClO2, HClO3, HClO4, which have the names hypochlorous, chlorous, chloric and perchloric acids. An acid is said to be monobasic, dibasic, tribasic, &c., according to the number of replaceable hydrogen atoms; thus HNO3 is monobasic, sulphuric acid H2SO4 dibasic, phosphoric acid H3PO4 tribasic.
An acid terminating in -ous forms a salt ending in -ite, and an oxyacid ending in -ic forms a salt ending in -ate. Thus the chlorine oxyacids enumerated above form salts named respectively hypochlorites, chlorites, chlorates and perchlorates. Salts formed from hydracids terminate in -ide, following the rule for binary compounds. An acid salt is one in which the whole amount of hydrogen has not been replaced by metal; a normal salt is one in which all the hydrogen has been replaced; and a basic salt is one in which part of the acid of the normal salt has been replaced by oxygen.
Chemical Formulae.—Opposite the name of each element in the second column of the above table, the symbol is given which is always employed to represent it. This symbol, however, not only represents the particular element, but a certain definite quantity of it. Thus, the letter H always stands for 1 atom or 1 part by weight of hydrogen, the letter N for 1 atom or 14 parts of nitrogen, and the symbol Cl for 1 atom or 35.5 parts of chlorine.8 Compounds are in like manner represented by writing the symbols of their constituent elements side by side, and if more than one atom of each element be present, the number is indicated by a numeral placed on the right of the symbol of the element either below or above the line. Thus, hydrochloric acid is represented by the formula HCl, that is to say, it is a compound of an atom of hydrogen with an atom of chlorine, or of 1 part by weight of hydrogen with 35.5 parts by weight of chlorine; again, sulphuric acid is represented by the formula H2SO4, which is a statement that it consists of 2 atoms of hydrogen, 1 of sulphur, and 4 of oxygen, and consequently of certain relative weights of these elements. A figure placed on the right of a symbol only affects the symbol to which it is attached, but when figures are placed in front of several symbols all are affected by it, thus 2H2SO4 means H2SO4 taken twice.
The distribution of weight in chemical change is readily expressed in the form of equations by the aid of these symbols; the equation
2HCl + Zn = ZnCl2 + H2,
for example, is to be read as meaning that from 73 parts of hydrochloric acid and 65 parts of zinc, 136 parts of zinc chloride and 2 parts of hydrogen are produced. The + sign is invariably employed in this way either to express combination or action upon, the meaning usually attached to the use of the sign = being that from such and such bodies such and such other bodies are formed.
Usually, when the symbols of the elements are written or printed with a figure to the right, it is understood that this indicates a molecule of the element, the symbol alone representing an atom. Thus, the symbols H2 and P4 indicate that the molecules of hydrogen and phosphorus respectively contain 2 and 4 atoms. Since, according to the molecular theory, in all cases of chemical change the action is between molecules, such symbols as these ought always to be employed. Thus, the formation of hydrochloric acid from hydrogen and chlorine is correctly represented by the equation
H2 + Cl2 = 2HCl;
that is to say, a molecule of hydrogen and a molecule of chlorine give rise to two molecules of hydrochloric acid; whilst the following equation merely represents the relative weights of the elements which enter into reaction, and is not a complete expression of what is supposed to take place:—
H + Cl = HCl.
In all cases it is usual to represent substances by formulae which to the best of our knowledge express their molecular composition in the state of gas, and not merely the relative number of atoms which they contain; thus, acetic acid consists of carbon, hydrogen and oxygen in the proportion of one atom of carbon, two of hydrogen, and one of oxygen, but its molecular weight corresponds to the formula C2H4O2, which therefore is always employed to represent acetic acid. When chemical change is expressed with the aid of molecular formulae not only is the distribution of weight represented, but by the mere inspection of the symbols it is possible to deduce from the law of gaseous combination mentioned above, the relative volumes which the agents and resultants occupy in the state of gas if measured at the same temperature and under the same pressure. Thus, the equation
2H2 + O2= 2H2O
not only represents that certain definite weights of hydrogen and oxygen furnish a certain definite weight of the compound which we term water, but that if the water in the state of gas, the hydrogen and the oxygen are all measured at the same temperature and pressure, the volume occupied by the oxygen is only half that occupied by the hydrogen, whilst the resulting water-gas will only occupy the same volume as the hydrogen. In other words, 2 volumes of oxygen and 4 volumes of hydrogen furnish 4 volumes of water-gas. A simple equation like this, therefore, when properly interpreted, affords a large amount of information. One other instance may be given; the equation
2NH3 = N2 + 3H2
represents the decomposition of ammonia gas into nitrogen and hydrogen gases by the electric spark, and it not only conveys the information that a certain relative weight of ammonia, consisting of certain relative weights of hydrogen and nitrogen, is broken up into certain relative weights of hydrogen and nitrogen, but also that the nitrogen will be contained in half the space which contained the ammonia, and that the volume of the hydrogen will be one and a half times as great as that of the original ammonia, so that in the decomposition of ammonia the volume becomes doubled.
Formulae which merely express the relative number of atoms of the different elements present in a compound are termed empirical formulae, and the formulae of all compounds whose molecular weights are undetermined are necessarily empirical. The molecular formula of a compound, however, is always a simple multiple of the empirical formula, if not identical with it; thus, the empirical formula of acetic acid is CH2O, and its molecular formula is C2H4O2, or twice CH2O. In addition to empirical and molecular formulae, chemists are in the habit of employing various kinds of rational formulae, called structural, constitutional or graphic formulae, &c., which not only express the molecular composition of the compounds to which they apply, but also embody certain assumptions as to the manner in which the constituent atoms are arranged, and convey more or less information with regard to the nature of the compound itself, viz. the class to which it belongs, the manner in which it is formed, and the behaviour it will exhibit under various circumstances. Before explaining these formulae it will be necessary, however, to consider the differences in combining power exhibited by the various elements.
Valency.—It is found that the number of atoms of a given element, of chlorine, for example, which unite with an atom of each of the other elements is very variable. Thus, hydrogen unites with but a single atom of chlorine, zinc with two, boron with three, silicon with four, phosphorus with five and tungsten with six. Those elements which are equivalent in combining or displacing power to a single atom of hydrogen are said to be univalent or monad elements; whilst those which are equivalent to two atoms of hydrogen are termed bivalent or dyad elements; and those equivalent to three, four, five or six atoms of hydrogen triad, tetrad, pentad or hexad elements. But not only is the combining power or valency (atomicity) of the elements different, it is also observed that one element may combine with another in several proportions, or that its valency may vary; for example, phosphorus forms two chlorides represented by the formulae PCl3 and PCl5, nitrogen the series of oxides represented by the formulae N2O, NO, (N2O3), N2O4, N2O5, molybdenum forms the chlorides MoCl2, MoCl3, MoCl4, MoCl5, MoCl6(?), and tungsten the chlorides WCl2, WCl4, WCl5, WCl6.
In explanation of these facts it is supposed that each element has a certain number of “units of affinity,” which may be entirely, or only in part, engaged when it enters into combination with other elements; and in those cases in which the entire number of units of affinity are not engaged by other elements, it is supposed that those which are thus disengaged neutralize each other, as it were. For example, in phosphorus pentachloride the five units of affinity possessed by the phosphorus atom are satisfied by the five monad atoms of chlorine, but in the trichloride two are disengaged, and, it may be supposed, satisfy each other. Compounds in which all the units of affinity of the contained elements are engaged are said to be saturated, whilst those in which the affinities of the contained elements are not all engaged by other elements are said to be unsaturated. According to this view, it is necessary to assume that, in all unsaturated compounds, two, or some even number of affinities are disengaged; and also that all elements which combine with an even number of monad atoms cannot combine with an odd number, and vice versa,—in other words, that the number of units of affinity active in the case of any given element must be always either an even or an odd number, and that it cannot be at one time an even and at another an odd number. There are, however, a few remarkable exceptions to this “law.” Thus, it must be supposed that in nitric oxide, NO, an odd number of affinities are disengaged, since a single atom of dyad oxygen is united with a single atom of nitrogen, which in all its compounds with other elements acts either as a triad or pentad. When nitric peroxide, N2O4, is converted into gas, it decomposes, and at about 180° C. its vapour entirely consists of molecules of the composition NO2; while at temperatures between this and 0° C. it consists of a mixture in different proportions of the two kinds of molecules, N2O4 and NO2. The oxide NO2 must be regarded as another instance of a compound in which an odd number of affinities of one of the contained elements are disengaged, since it contains two atoms of dyad oxygen united with a single atom of triad or pentad nitrogen. Again, when tungsten hexachloride is converted into vapour it is decomposed into chlorine and a pentachloride, having a normal vapour density, but as in the majority of its compounds tungsten acts as a hexad, we apparently must regard its pentachloride as a compound in which an odd number of free affinities are disengaged. Hitherto no explanation has been given of these exceptions to what appears to be a law of almost universal application, viz. that the sum of the units of affinity of all the atoms in a compound is an even number.
The number of units of affinity active in the case of any particular element is largely dependent, however, upon the nature of the element or elements with which it is associated. Thus, an atom of iodine only combines with one of hydrogen, but may unite with three of chlorine, which never combines with more than a single atom of hydrogen; an atom of phosphorus unites with only three atoms of hydrogen, but with five of chlorine, or with four of hydrogen and one of iodine; and the chlorides corresponding to the higher oxides of lead, nickel, manganese and arsenic, PbO2, Ni2O3, MnO2 and AS2O5 do not exist as stable compounds, but the lower chlorides, PbCl2, NiCl2, MnCl2 and AsCl3, are very stable.
The valency of an element is usually expressed by dashes or Roman numerals placed on the right of its symbol, thus: H′, O′′, B′′′, CIV, PV, MoVI; but in constructing graphic formulae the symbols of the elements are written with as many lines attached to each symbol as the element which it represents has units of affinity.
The periodic law (see Element) permits a grouping of the elements according to their valency as follows:—Group O.: helium, neon, argon, krypton and xenon appear to be devoid of valency. Group I.: the alkali metals Li, Na, K, Rb, Cs, and also Ag, monovalent; Cu, monovalent and divalent; Au, monovalent and trivalent. Group II.: the alkaline earth metals Ca, Sr, Ba, and also Be (Gl), Mg, Zn, Cd, divalent; Hg, monovalent and divalent. Group III.: B, trivalent; Al, trivalent, but possibly also tetra- or penta-valent; Ga, divalent and trivalent; In, mono-, di- and tri-valent; Tl, monovalent and trivalent. Group IV.: C, Si, Ge, Zr, Th, tetravalent; Ti, tetravalent and hexavalent; Sn, Pb, divalent and tetravalent; Ce, trivalent and tetravalent. Group V.: N, trivalent and pentavalent, but divalent in nitric oxide; P, As, Sb, Bi, trivalent and pentavalent, the last being possibly divalent in BiO and BiCl2. Group VI.: O, usually divalent, but tetravalent and possibly hexavalent in oxonium and other salts; S, Se, Te, di-, tetra- and hexa-valent; Cr, di-, tri- and hexa-valent; Mo, W, di-, tri-, tetra-, penta- and hexa-valent. Group VII.: H(?), monovalent; the halogens F, Cl, Br, I, usually monovalent, but possibly also tri- and pentavalent; Mn, divalent and trivalent, and possibly heptavalent in permanganates. Group VIII.: Fe, Co, divalent and trivalent; Ni, divalent; Os, Ru, hexavalent and octavalent; Pd, Pt, divalent and tetravalent; Ir, tri-, tetra- and hexa-valent. (See also Valency.)
Constitutional Formulae.—Graphic or constitutional formulae are employed to express the manner in which the constituent atoms of compounds are associated together; for example, the trioxide of sulphur is usually regarded as a compound of an atom of hexad sulphur with three atoms of dyad oxygen, and this hypothesis is illustrated by the graphic formula
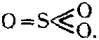
When this oxide is brought into contact with water it combines with it forming sulphuric acid, H2SO4.
In this compound only two of the oxygen atoms are wholly associated with the sulphur atom, each of the remaining oxygen atoms being united by one of its affinities to the sulphur atoms, and by the remaining affinity to an atom of hydrogen; thus—
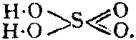
The graphic formula of a sulphate is readily deduced by remembering that the hydrogen atoms are partially or entirely replaced. Thus acid sodium sulphate, normal sodium sulphate, and zinc sulphate have the formulae

Again, the reactions of acetic acid, C2H4O2, show that the four atoms of hydrogen which it contains have not all the same function, and also that the two atoms of oxygen have different functions; the graphic formula which we are led to assign to acetic acid, viz.
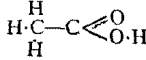
serves in a measure to express this, three of the atoms of hydrogen being represented as associated with one of the atoms of carbon, whilst the fourth atom is associated with an atom of oxygen which is united by a single affinity to the second atom of carbon, to which, however, the second atom of oxygen is united by both of its affinities. It is not to be supposed that there are any actual bonds of union between the atoms; graphic formulae such as these merely express the hypothesis that certain of the atoms in a compound come directly within the sphere of attraction of certain other atoms, and only indirectly within the sphere of attraction of others,—an hypothesis to which chemists are led by observing that it is often possible to separate a group of elements from a compound, and to displace it by other elements or groups of elements.
Rational formulae of a much simpler description than these graphic formulae are generally employed. For instance, sulphuric acid is usually represented by the formula SO2(OH)2, which indicates that it may be regarded as a compound of the group SO2 with twice the group OH. Each of these OH groups is equivalent in combining or displacing power to a monad element, since it consists of an atom of dyad oxygen associated with a single atom of monad hydrogen, so that in this case the SO2 group is equivalent to an atom of a dyad element. This formula for sulphuric acid, however, merely represents such facts as that it is possible to displace an atom of hydrogen and an atom of oxygen in sulphuric acid by a single atom of chlorine, thus forming the compound SO3HCl; and that by the action of water on the compound SO2Cl2 twice the group OH, or water minus an atom of hydrogen, is introduced in place of the two monad atoms of chlorine—
SO2Cl2 + 2HOH = SO2(OH)2 + 2HCl.
Water. Sulphuric acid.
Constitutional formulae like these, in fact, are nothing more than symbolic expressions of the character of the compounds which they represent, the arrangement of symbols in a certain definite manner being understood to convey certain information with regard to the compounds represented.
Groups of two or more atoms like SO2 and OH, which are capable of playing the part of elementary atoms (that is to say, which can be transferred from compound to compound), are termed compound radicals, the elementary atoms being simple radicals. Thus, the atom of hydrogen is a monad simple radical, the atom of oxygen a dyad simple radical, whilst the group OH is a monad compound radical.
It is often convenient to regard compounds as formed upon certain types; alcohol, for example, may be said to be a compound formed upon the water type, that is to say, a compound formed from water by displacing one of the atoms of hydrogen by the group of elements C2H5, thus—
| O | { | H | O | { | C2H5 |
| H | H | ||||
| Water | Alcohol. | ||||
Constitutional formulae become of preponderating importance when we consider the more complicated inorganic and especially organic compounds. Their full significance is treated in the section of this article dealing with organic chemistry, and in the articles Isomerism and Stereo-isomerism.
Chemical Action.—Chemical change or chemical action may be said to take place whenever changes occur which involve an alteration in the composition of molecules, and may be the result of the action of agents such as heat, electricity or light, or of two or more elements or compounds upon each other.
Three kinds of changes are to be distinguished, viz. changes which involve combination, changes which involve decomposition or separation, and changes which involve at the same time both decomposition and combination. Changes of the first and second kind, according to our views of the constitution of molecules, are probably of very rare occurrence; in fact, chemical action appears almost always to involve the occurrence of both these kinds of change, for, as already pointed out, we must assume that the molecules of hydrogen, oxygen and several other elements are diatomic, or that they consist of two atoms. Indeed, it appears probable that with few exceptions the elements are all compounds of similar atoms united together by one or more units of affinity, according to their valencies. If this be the case, however, it is evident that there is no real distinction between the reactions which take place when two elements combine together and when an element in a compound is displaced by another. The combination, as it is ordinarily termed, of chlorine with hydrogen, and the displacement of iodine in potassium iodide by the action of chlorine, may be cited as examples; if these reactions are represented, as such reactions very commonly are, by equations which merely express the relative weights of the bodies which, enter, into reaction, and of the products, thus—
| H | + | Cl | = | HCl |
| Hydrogen. | Chlorine. | Hydrochloric acid. |
| KI | + | Cl | = | KCl | + | I |
| Potassium iodide. | Chlorine. | Potassium chloride. | Iodine. |
they appear to differ in character; but if they are correctly represented by molecular equations, or equations which express the relative number of molecules which enter into reaction and which result from the reaction, it will be obvious that the character of the reaction is substantially the same in both cases, and that both are instances of the occurrence of what is ordinarily termed double decomposition—
| H2 | + | Cl2 | = | 2HCl |
| Hydrogen. | Chlorine. | Hydrochloric acid. |
| 2KI | + | Cl2 | = | 2KCl | + | I2. |
| Potassium iodide. | Chlorine. | Potassium chloride. | Iodine. |
In all cases of chemical change energy in the form of heat is either developed or absorbed, and the amount of heat developed or absorbed in a given reaction is as definite as are the weights of the substance engaged in the reaction. Thus, in the production of hydrochloric acid from hydrogen and chlorine 22,000 calories are developed; in the production of hydrobromic acid from hydrogen and bromine, however, only 8440 calories are developed; and in the formation of hydriodic acid from hydrogen and iodine 6040 calories are absorbed.
This difference in behaviour of the three elements, chlorine, bromine and iodine, which in many respects exhibit considerable resemblance, may be explained in the following manner. We may suppose that in the formation of gaseous hydrochloric acid from gaseous chlorine and hydrogen, according to the equation
H2 + Cl2 = HCl + HCl,
a certain amount of energy is expended in separating the atoms of hydrogen in the hydrogen molecule, and the atoms of chlorine in the chlorine molecule, from each other; but that heat is developed by the combination of the hydrogen atoms with the chlorine atoms, and that, as more energy is developed by the union of the atoms of hydrogen and chlorine than is expended in separating the hydrogen atoms from each other and the chlorine atoms from one another, the result of the action of the two elements upon each other is the development of heat,—the amount finally developed in the reaction being the difference between that absorbed in decomposing the elementary molecules and that developed by the combination of the atoms of chlorine and hydrogen. In the formation of gaseous hydrobromic acid from liquid bromine and gaseous hydrogen—
H2 + Br2 = HBr + HBr,
in addition to the energy expended in decomposing the hydrogen and bromine molecules, energy is also expended in converting the liquid bromine into the gaseous condition, and probably less heat is developed by the combination of bromine and hydrogen than by the combination of chlorine and hydrogen, so that the amount of heat finally, developed is much less than is developed in the formation of hydrochloric acid. Lastly, in the production of gaseous hydriodic acid from hydrogen and solid iodine—
H2 + I2 = HI + HI,
so much energy is expended in the decomposition of the hydrogen and iodine molecules and in the conversion of the iodine into the gaseous condition, that the heat which it may be supposed is developed by the combination of the hydrogen and iodine atoms is insufficient to balance the expenditure, and the final result is therefore negative; hence it is necessary in forming hydriodic acid from its elements to apply heat continuously.
These compounds also afford examples of the fact that, generally speaking, those compounds are most readily formed, and are most stable, in the formation of which the most heat is developed. Thus, chlorine enters into reaction with hydrogen, and removes hydrogen from hydrogenized bodies, far more readily than bromine; and hydrochloric acid is a far more stable substance than hydrobromic acid, hydriodic acid being greatly inferior even to hydrobromic acid in stability. Compounds formed with the evolution of heat are termed exothermic, while those formed with an absorption are termed endothermic. Explosives are the commonest examples of endothermic compounds.
When two substances which by their action upon each other develop much heat enter into reaction, the reaction is usually complete without the employment of an excess of either; for example, when a mixture of hydrogen and oxygen, in the proportions to form water—
2H2 + O2 = 2OH2,
is exploded, it is entirely converted into water. This is also the case if two substances are brought together in solution, by the action of which upon each other a third body is formed which is insoluble in the solvent employed, and which also does not tend to react upon any of the substances present; for instance, when a solution of a chloride is added to a solution of a silver salt, insoluble silver chloride is precipitated, and almost the whole of the silver is removed from solution, even if the amount of the chloride employed be not in excess of that theoretically required.
But if there be no tendency to form an insoluble compound, Or one which is not liable to react upon any of the other substances present, this is no longer the case. For example, when a solution of a ferric salt is added to a solution of potassium thiocyanate, a deep red coloration is produced, owing to the formation of ferric thiocyanate. Theoretically the reaction takes place in the case of ferric nitrate in the manner represented by the equation
| Fe(NO3)3 | + | 3KCNS | = | Fe(CNS)3 | + | 3KNO3; |
| Ferric nitrate. | Potassium thiocyanate. | Ferric thiocyanate. | Potassium nitrate. |
but it is found that even when more than sixty times the amount of potassium thiocyanate required by this equation is added, a portion of the ferric nitrate still remains unconverted, doubtless owing to the occurrence of the reverse change—
Fe(CNS)3 + 3KNO3 = Fe(NO3)3 + 3KCNS.
In this, as in most other cases in which substances act upon one another under such circumstances that the resulting compounds are free to react, the extent to which the different kinds of action which may occur take place is dependent upon the mass of the substances present in the mixture. As another instance of this kind, the decomposition of bismuth chloride by water may be cited. If a very large quantity of water be added, the chloride is entirely decomposed in the manner represented by the equation—
| BiCl3 | + | OH2 | = | BiOCl | + | 2HCl, |
| Bismuth chloride. | Bismuth oxychloride. |
the oxychloride being precipitated; but if smaller quantities of water be added the decomposition is incomplete, and it is found that the extent to which decomposition takes place is proportional to the quantity of water employed, the decomposition being incomplete, except in presence of large quantities of water, because of the occurrence of the reverse action—
BiOCl + 2HCl = BiCl3 + OH2.
Chemical change which merely involves simple decomposition is thus seen to be influenced by the masses of the reacting substances and the presence of the products of decomposition; in other words the system of reacting substances and resultants form a mixture in which chemical action has apparently ceased, or the system is in equilibrium. Such reactions are termed reversible (see Chemical Action).
III. Inorganic Chemistry
Inorganic chemistry is concerned with the descriptive study of the elements and their compounds, except those of carbon. Reference should be made to the separate articles on the different elements and the more important compounds for their preparation, properties and uses. In this article the development of this branch of the science is treated historically.
The earliest discoveries in inorganic chemistry are to be found in the metallurgy, medicine and chemical arts of the ancients. The Egyptians obtained silver, iron, copper, lead, zinc and tin, either pure or as alloys, by smelting the ores; mercury is mentioned by Theophrastus (c. 300 B.C.). The manufacture of glass, also practised in Egypt, demanded a knowledge of sodium or potassium carbonates; the former occurs as an efflorescence on the shores of certain lakes; the latter was obtained from wood ashes. Many substances were used as pigments: Pliny records white lead, cinnabar, verdigris and red oxide of iron; and the preparation of coloured glasses and enamels testifies to the uses to which these and other substances were put. Salts of ammonium were also known; while alum was used as a mordant in dyeing. Many substances were employed in ancient medicine: galena was the basis of a valuable Egyptian cosmetic and drug; the arsenic sulphides, realgar and orpiment, litharge, alum, saltpetre, iron rust were also used. Among the Arabian and later alchemists we find attempts made to collate compounds by specific properties, and it is to these writers that we are mainly indebted for such terms as “alkali,” “sal,” &c. The mineral acids, hydrochloric, nitric and sulphuric acids, and also aqua regia (a mixture of hydrochloric and nitric acids) were discovered, and the vitriols, alum, saltpetre, sal-ammoniac, ammonium carbonate, silver nitrate (lunar caustic) became better known. The compounds of mercury attracted considerable attention, mainly on account of their medicinal properties; mercuric oxide and corrosive sublimate were known to pseudo-Geber, and the nitrate and basic sulphate to “Basil Valentine.” Antimony and its compounds formed the subject of an elaborate treatise ascribed to this last writer, who also contributed to our knowledge of the compounds of zinc, bismuth and arsenic. All the commonly occurring elements and compounds appear to have received notice by the alchemists; but the writings assigned to the alchemical period are generally so vague and indefinite that it is difficult to determine the true value of the results obtained.
In the succeeding iatrochemical period, the methods of the alchemists were improved and new ones devised. Glauber showed how to prepare hydrochloric acid, spiritus salis, by heating rock-salt with sulphuric acid, the method in common use to-day; and also nitric acid from saltpetre and arsenic trioxide. Libavius obtained sulphuric acid from many substances, e.g. alum, vitriol, sulphur and nitric acid, by distillation. The action of these acids on many metals was also studied; Glauber obtained zinc, stannic, arsenious and cuprous chlorides by dissolving the metals in hydrochloric acid, compounds hitherto obtained by heating the metals with corrosive sublimate, and consequently supposed to contain mercury. The scientific study of salts dates from this period, especial interest being taken in those compounds which possessed a medicinal or technical value. In particular, the salts of potassium, sodium and ammonium were carefully investigated, but sodium and potassium salts were rarely differentiated.9 The metals of the alkaline-earths were somewhat neglected; we find Georg Agricola considering gypsum (calcium sulphate) as a compound of lime, while calcium nitrate and chloride became known at about the beginning of the 17th century. Antimonial, bismuth and arsenical compounds were assiduously studied, a direct consequence of their high medicinal importance; mercurial and silver compounds were investigated for the same reason. The general tendency of this period appears to have taken the form of improving and developing the methods of the alchemists; few new fields were opened, and apart from a more complete knowledge of the nature of salts, no valuable generalizations were attained.
The discovery of phosphorus by Brand, a Hamburg alchemist, in 1669 excited chemists to an unwonted degree; it was also independently prepared by Robert Boyle and J. Kunckel, Brand having kept his process secret. Towards the middle of the 18th century two new elements were isolated: cobalt by G. Brandt in 1742, and nickel by A.F. Cronstedt in 1750. These discoveries were followed by Daniel Rutherford’s isolation of nitrogen in 1772, and by K. Scheele’s isolation of chlorine and oxygen in 1774 (J. Priestley discovered oxygen independently at about the same time), and his investigation of molybdic and tungstic acids in the following year; metallic molybdenum was obtained by P.J. Hjelm in 1783, and tungsten by Don Fausto d’Elhuyar; manganese was isolated by J.G. Gahn in 1774. In 1784 Henry Cavendish thoroughly examined hydrogen, establishing its elementary nature; and he made the far-reaching discovery that water was composed of two volumes of hydrogen to one of oxygen.
The phlogistic theory, which pervaded the chemical doctrine of this period, gave rise to continued study of the products of calcination and combustion; it thus happened that the knowledge of oxides and oxidation products was considerably developed. The synthesis of nitric acid by passing electric sparks through moist air by Cavendish is a famous piece of experimental work, for in the first place it determined the composition of this important substance, and in the second place the minute residue of air which would not combine, although ignored for about a century, was subsequently examined by Lord Rayleigh and Sir William Ramsay, who showed that it consists of a mixture of elementary substances—argon, krypton, neon and xenon (see Argon).
The 18th century witnessed striking developments in pneumatic chemistry, or the chemistry of gases, which had been begun by van Helmont, Mayow, Hales and Boyle. Gases formerly considered to be identical came to be clearly distinguished, and many new ones were discovered. Atmospheric air was carefully investigated by Cavendish, who showed that it consisted of two elementary constituents: nitrogen, which was isolated by Rutherford in 1772, and oxygen, isolated in 1774; and Black established the presence, in minute quantity, of carbon dioxide (van Helmont’s gas sylvestre). Of the many workers in this field, Priestley occupies an important position. A masterly device, initiated by him, was to collect gases over mercury instead of water; this enabled him to obtain gases previously only known in solution, such as ammonia, hydrochloric acid, silicon fluoride and sulphur dioxide. Sulphuretted hydrogen and nitric oxide were discovered at about the same time.
Returning to the history of the discovery of the elements and their more important inorganic compounds, we come in 1789 to M.H. Klaproth’s detection of a previously unknown constituent of the mineral pitchblende. He extracted a substance to which he assigned the character of an element, naming it uranium (from Οὐρανός, heaven); but it was afterwards shown by E.M. Péligot, who prepared the pure metal, that Klaproth’s product was really an oxide. This element was investigated at a later date by Sir Henry Roscoe, and more thoroughly and successfully by C. Zimmermann and Alibegoff. Pitchblende attained considerable notoriety towards the end of the 19th century on account of two important discoveries. The first, made by Sir William Ramsay in 1896, was that the mineral evolved a peculiar gas when treated with sulphuric acid; this gas, helium (q.v.), proved to be identical with a constituent of the sun’s atmosphere, detected as early as 1868 by Sir Norman Lockyer during a spectroscopic examination of the sun’s chromosphere. The second discovery, associated with the Curies, is that of the peculiar properties exhibited by the impure substance, and due to a constituent named radium. The investigation of this substance and its properties (see Radioactivity) has proceeded so far as to render it probable that the theory of the unalterability of elements, and also the hitherto accepted explanations of various celestial phenomena—the source of solar energy and the appearances of the tails of comets—may require recasting.
In the same year as Klaproth detected uranium, he also isolated zirconia or zirconium oxide from the mineral variously known as zircon, hyacinth, jacynth and jargoon; but he failed to obtain the metal, this being first accomplished some years later by Berzelius, who decomposed the double potassium zirconium fluoride with potassium. In the following year, 1795, Klaproth announced the discovery of a third new element, titanium; its isolation (in a very impure form), as in the case of zirconium, was reserved for Berzelius.
Passing over the discovery of carbon disulphide by W.A. Lampadius in 1796, of chromium by L.N. Vauquelin in 1797, and Klaproth’s investigation of tellurium in 1798, the next important series of observations was concerned with platinum and the allied metals. Platinum had been described by Antonio de Ulloa in 1748, and subsequently discussed by H.T. Scheffer in 1752. In 1803 W.H. Wollaston discovered palladium, especially interesting for its striking property of absorbing (“occluding”) as much as 376 volumes of hydrogen at ordinary temperatures, and 643 volumes at 90°. In the following year he discovered rhodium; and at about the same time Smithson Tennant added two more to the list—iridium and osmium; the former was so named from the changing tints of its oxides (ῒρις, rainbow), and the latter from the odour of its oxide (ὀσμή, smell). The most recently discovered “platinum metal,” ruthenium, was recognized by C.E. Claus in 1845. The great number and striking character of the compounds of this group of metals have formed the subject of many investigations, and already there is a most voluminous literature. Berzelius was an early worker in this field; he was succeeded by Bunsen, and Deville and Debray, who worked out the separation of rhodium; and at a later date by P.T. Cleve, the first to make a really thorough study of these elements and their compounds. Of especial note are the curious compounds formed by the union of carbon monoxide with platinous chloride, discovered by Paul Schützenberger and subsequently investigated by F.B. Mylius and F. Foerster and by Pullinger; the phosphoplatinic compounds formed primarily from platinum and phosphorus pentachloride; and also the “ammino” compounds, formed by the union of ammonia with the chloride, &c., of these metals, which have been studied by many chemists, especially S.M. Jörgensen. Considerable uncertainty existed as to the atomic weights of these metals, the values obtained by Berzelius being doubtful. K.F.O. Seubert redetermined this constant for platinum, osmium and iridium; E.H. Keiser for palladium, and A.A. Joly for ruthenium.
The beginning of the 19th century witnessed the discovery of certain powerful methods for the analysis of compounds and the isolation of elements. Berzelius’s investigation of the action of the electric current on salts clearly demonstrated the invaluable assistance that electrolysis could render to the isolator of elements; and the adoption of this method by Sir Humphry Davy for the analysis of the hydrates of the metals of the alkalis and alkaline earths, and the results which he thus achieved, established its potency. In 1808 Davy isolated sodium and potassium; he then turned his attention to the preparation of metallic calcium, barium, strontium and magnesium. Here he met with greater difficulty, and it is to be questioned whether he obtained any of these metals even in an approximately pure form (see Electrometallurgy). The discovery of boron by Gay Lussac and Davy in 1809 led Berzelius to investigate silica (silex). In the following year he announced that silica was the oxide of a hitherto unrecognized element, which he named silicium, considering it to be a metal. This has proved to be erroneous; it is non-metallic in character, and its name was altered to silicon, from analogy with carbon and boron. At the same time Berzelius obtained the element, in an impure condition, by fusing silica with charcoal and iron in a blast furnace; its preparation in a pure condition he first accomplished in 1823, when he invented the method of heating double potassium fluorides with metallic potassium. The success which attended his experiments in the case of silicon led him to apply it to the isolation of other elements. In 1824 he obtained zirconium from potassium zirconium fluoride; the preparation of (impure) titanium quickly followed, and in 1828 he obtained thorium. A similar process, and equally efficacious, was introduced by F. Wöhler in 1827. It consisted in heating metallic chlorides with potassium, and was first applied to aluminium, which was isolated in 1827; in the following year, beryllium chloride was analysed by the same method, beryllium oxide (berylla or glucina) having been known since 1798, when it was detected by L. N. Vauquelin in the gem-stone beryl.
In 1812 B. Courtois isolated the element iodine from “kelp,” the burnt ashes of marine plants. The chemical analogy of this substance to chlorine was quickly perceived, especially after its investigation by Davy and Gay Lussac. Cyanogen, a compound which in combination behaved very similarly to chlorine and iodine, was isolated in 1815 by Gay Lussac. This discovery of the first of the then-styled “compound radicals” exerted great influence on the prevailing views of chemical composition. Hydrochloric acid was carefully investigated at about this time by Davy, Faraday and Gay Lussac, its composition and the elementary nature of chlorine being thereby established.
In 1817 F. Stromeyer detected a new metallic element, cadmium, in certain zinc ores; it was rediscovered at subsequent dates by other observers and its chemical resemblance to zinc noticed. In the same year Berzelius discovered selenium in a deposit from sulphuric acid chambers, his masterly investigation including a study of the hydride, oxides and other compounds. Selenic acid was discovered by E. Mitscherlich, who also observed the similarity of the crystallographic characters of selenates and sulphates, which afforded valuable corroboration of his doctrine of isomorphism. More recent and elaborate investigations in this direction by A.E.H. Tutton have confirmed this view.
In 1818 L.J. Thénard discovered hydrogen dioxide, one of the most interesting inorganic compounds known, which has since been carefully investigated by H.E. Schöne, M. Traube, Wolfenstein and others. About the same time, J.A. Arfvedson, a pupil of Berzelius, detected a new element, which he named lithium, in various minerals—notably petalite. Although unable to isolate the metal, he recognized its analogy to sodium and potassium; this was confirmed by R. Bunsen and A. Matthiessen in 1855, who obtained the metal by electrolysis and thoroughly examined it and its compounds. Its crimson flame-coloration was observed by C.G. Gmelin in 1818.
The discovery of bromine in 1826 by A.J. Balard completed for many years Berzelius’s group of “halogen” elements; the remaining member, fluorine, notwithstanding many attempts, remained unisolated until 1886, when Henri Moissan obtained it by the electrolysis of potassium fluoride dissolved in hydrofluoric acid. Hydrobromic and hydriodic acids were investigated by Gay Lussac and Balard, while hydrofluoric acid received considerable attention at the hands of Gay Lussac, Thénard and Berzelius. We may, in fact, consider that the descriptive study of the various halogen compounds dates from about this time. Balard discovered chlorine monoxide in 1834, investigating its properties and reactions; and his observations on hypochlorous acid and hypochlorites led him to conclude that “bleaching-powder” or “chloride of lime” was a compound or mixture in equimolecular proportions of calcium chloride and hypochlorite, with a little calcium hydrate. Gay Lussac investigated chloric acid; Stadion discovered perchloric acid, since more fully studied by G.S. Serullas and Roscoe; Davy and Stadion investigated chlorine peroxide, formed by treating potassium chlorate with sulphuric acid. Davy also described and partially investigated the gas, named by him “euchlorine,” obtained by heating potassium chlorate with hydrochloric acid; this gas has been more recently examined by Pebal. The oxy-acids of iodine were investigated by Davy and H.G. Magnus; periodic acid, discovered by the latter, is characterized by the striking complexity of its salts as pointed out by Kimmins.
In 1830 N.G. Sefström definitely proved the existence of a metallic element vanadium, which had been previously detected (in 1801) in certain lead ores by A.M. del Rio; subsequent elaborate researches by Sir Henry Roscoe showed many inaccuracies in the conclusions of earlier workers (for instance, the substance considered to be the pure element was in reality an oxide) and provided science with an admirable account of this element and its compounds. B.W. Gerland contributed to our knowledge of vanadyl salts and the vanadic acids. Chemically related to vanadium are the two elements tantalum and columbium or niobium. These elements occur in the minerals columbite and tantalite, and their compounds became known in the early part of the 19th century by the labours of C. Hatchett, A.G. Ekeberg, W.H. Wollaston and Berzelius. But the knowledge was very imperfect; neither was it much clarified by H. Rose, who regarded niobium oxide as the element. The subject was revived in 1866 by C.W. Blomstrand and J.C. Marignac, to whom is due the credit of first showing the true chemical relations of these elements. Subsequent researches by Sainte Claire Deville and L.J. Troost, and by A.G. Krüss and L.E. Nilson, and subsequently (1904) by Hall, rendered notable additions to our knowledge of these elements and their compounds. Tantalum has in recent years been turned to economic service, being employed, in the same manner as tungsten, for the production of the filaments employed in incandescent electric lighting.In 1833 Thomas Graham, following the paths already traced out by E.D. Clarke, Gay Lussac and Stromeyer, published his masterly investigation of the various phosphoric acids and their salts, obtaining results subsequently employed by J. von Liebig in establishing the doctrine of the characterization and basicity of acids. Both phosphoric and phosphorous acids became known, although imperfectly, towards the end of the 18th century; phosphorous acid was first obtained pure by Davy in 1812, while pure phosphorous oxide, the anhydride of phosphorous acid, remained unknown until T.E. Thorpe’s investigation of the products of the slow combustion of phosphorus. Of other phosphorus compounds we may here notice Gengembre’s discovery of phosphuretted hydrogen (phosphine) in 1783, the analogy of which to ammonia was first pointed out by Davy and supported at a later date by H. Rose; liquid phosphuretted hydrogen was first obtained by Thénard in 1838; and hypophosphorous acid was discovered by Dulong in 1816. Of the halogen compounds of phosphorus, the trichloride was discovered by Gay Lussac and Thénard, while the pentachloride was obtained by Davy. The oxychloride, bromides, and other compounds were subsequently discovered; here we need only notice Moissan’s preparation of the trifluoride and Thorpe’s discovery of the pentafluoride, a compound of especial note, for it volatilizes unchanged, giving a vapour of normal density and so demonstrating the stability of a pentavalent phosphorus compound (the pentachloride and pentabromide dissociate into a molecule of the halogen element and phosphorus trichloride).
In 1840 C.F. Schönbein investigated ozone, a gas of peculiar odour (named from the Gr. ὄζειν, to smell) observed in 1785 by Martin van Marum to be formed by the action of a silent electric discharge on the oxygen of the air; he showed it to be an allotropic modification of oxygen, a view subsequently confirmed by Marignac, Andrews and Soret. In 1845 a further contribution to the study of allotropy was made by Anton Schrötter, who investigated the transformations of yellow and red phosphorus, phenomena previously noticed by Berzelius, the inventor Of the term “allotropy.” The preparation of crystalline boron in 1856 by Wöhler and Sainte Claire Deville showed that this element also existed in allotropic forms, amorphous boron having been obtained simultaneously and independently in 1809 by Gay Lussac and Davy. Before leaving this phase of inorganic chemistry, we may mention other historical examples of allotropy. Of great importance is the chemical identity of the diamond, graphite and charcoal, a fact demonstrated in part by Lavoisier in 1773, Smithson Tennant in 1706, and by Sir George Steuart-Mackenzie (1780-1848), who showed that equal weights of these three substances yielded the same weight of carbon dioxide on combustion. The allotropy of selenium was first investigated by Berzelius; and more fully in 1851 by J.W. Hittorf, who carefully investigated the effects produced by heat; crystalline selenium possesses a very striking property, viz. when exposed to the action of light its electric conductivity increases. Another element occurring in allotropic forms is sulphur, of which many forms have been described. E. Mitscherlich was an early worker in this field. A modification known as “black sulphur,” soluble in water, was announced by F.L. Knapp in 1848, and a colloidal modification was described by H. Debus. The dynamical equilibrium between rhombic, liquid and monosymmetric sulphur has been worked out by H.W. Bakhuis Roozeboom. The phenomenon of allotropy is not confined to the non-metals, for evidence has been advanced to show that allotropy is far commoner than hitherto supposed. Thus the researches of Carey Lea, E.A. Schneider and others, have proved the existence of “colloidal silver”; similar forms of the metals gold, copper, and of the platinum metals have been described. The allotropy of arsenic and antimony is also worthy of notice, but in the case of the first element the variation is essentially non-metallic, closely resembling that of phosphorus. The term allotropy has also been applied to inorganic compounds, identical in composition, but assuming different crystallographic forms. Mercuric oxide, sulphide and iodide; arsenic trioxide; titanium dioxide and silicon dioxide may be cited as examples.
The joint discovery in 1859 of the powerful method of spectrum analysis (see Spectroscopy) by G.R. Kirchhoff and R.W. Bunsen, and its application to the detection and the characterization of elements when in a state of incandescence, rapidly led to the discovery of many hitherto unknown elements. Within two years of the invention the authors announced the discovery of two metals, rubidium and caesium, closely allied to sodium, potassium and lithium in properties, in the mineral lepidolite and in the Dürkheim mineral water. In 1861 Sir William Crookes detected thallium (named from the Gr. θάλλος, a green bud, on account of a brilliant green line in its spectrum) in the selenious mud of the sulphuric acid manufacture; the chemical affinities of this element, on the one hand approximating to the metals of the alkalis, and on the other hand to lead, were mainly established by C.A. Lamy. Of other metals first detected by the spectroscope mention is to be made of indium, determined by F. Reich and H.T. Richter in 1863, and of gallium, detected in certain zinc blendes by Lecoq de Boisbaudran in 1875. The spectroscope has played an all-important part in the characterization of the elements, which, in combination with oxygen, constitute the group of substances collectively named the “rare earths.” The substances occur, in very minute quantity, in a large number of sparingly-distributed and comparatively rare minerals—euxenite, samarksite, cerite, yttrotantalite, &c. Scandinavian specimens of these minerals were examined by J. Gadolin, M.H. Klaproth, and especially by Berzelius; these chemists are to be regarded as the pioneers in this branch of descriptive chemistry. Since their day many chemists have entered the lists, new and powerful methods of research have been devised, and several new elements definitely characterized. Our knowledge on many points, however, is very chaotic; great uncertainty and conflict of evidence circulate around many of the “new elements” which have been announced, so much so that P.T. Cleve proposed to divide the “rare earth” metals into two groups, (1) “perfectly characterized”; (2) “not yet thoroughly characterized.” The literature of this subject is very large. The memorial address on J.C.G. de Marignac, a noted worker in this field, delivered by Cleve, a high authority on this subject, before the London Chemical Society (J.C.S. Trans., 1895, p. 468), and various papers in the same journal by Sir William Crookes, Bohuslav Brauner and others should be consulted for details.
In the separation of the constituents of the complex mixture of oxides obtained from the “rare earth” minerals, the methods generally forced upon chemists are those of fractional precipitation or crystallization; the striking resemblances of the compounds of these elements rarely admitting of a complete separation by simple precipitation and filtration. The extraordinary patience requisite to a successful termination of such an analysis can only be adequately realized by actual research; an idea may be obtained from Crookes’s Select Methods in Analysis. Of recent years the introduction of various organic compounds as precipitants or reagents has reduced the labour of the process; and advantage has also been taken of the fairly complex double salts which these metals form with compounds. The purity of the compounds thus obtained is checked by spectroscopic observations. Formerly the spark- and absorption-spectra were the sole methods available; a third method was introduced by Crookes, who submitted the oxides, or preferably the basic sulphates, to the action of a negative electric discharge in vacuo, and investigated the phosphorescence induced spectroscopically. By such a study in the ultra-violet region of a fraction prepared from crude yttria he detected a new element victorium, and subsequently by elaborate fractionation obtained the element itself.
The first earth of this group to be isolated (although in an impure form) was yttria, obtained by Gadolin in 1794 from the mineral gadolinite, which was named after its discoverer and investigator. Klaproth and Vauquelin also investigated this earth, but without detecting that it was a complex mixture—a discovery reserved for C.G. Mosander. The next discovery, made independently and simultaneously in 1803 by Klaproth and by W. Hisinger and Berzelius, was of ceria, the oxide of cerium, in the mineral cerite found at Ridderhytta, Westmannland, Sweden. These crude earths, yttria and ceria, have supplied most if not all of the “rare earth” metals. In 1841 Mosander, having in 1839 discovered a new element lanthanum in the mineral cerite, isolated this element and also a hitherto unrecognized substance, didymia, from crude yttria, and two years later he announced the determination of two fresh constituents of the same earth, naming them erbia and terbia. Lanthanum has retained its elementary character, but recent attempts at separating it from didymia have led to the view that didymium is a mixture of two elements, praseodymium and neodymium (see Didymium). Mosander’s erbia has been shown to contain various other oxides—thulia, holmia, &c.—but this has not yet been perfectly worked out. In 1878 Marignac, having subjected Mosander’s erbia, obtained from gadolinite, to a careful examination, announced the presence of a new element, ytterbium; this discovery was confirmed by Nilson, who in the following year discovered another element, scandium, in Marignac’s ytterbia. Scandium possesses great historical interest, for Cleve showed that it was one of the elements predicted by Mendeléeff about ten years previously from considerations based on his periodic classification of the elements (see Element). Other elements predicted and characterized by Mendeléeff which have been since realized are gallium, discovered in 1875, and germanium, discovered in 1885 by Clemens Winkler.
In 1880 Marignac examined certain earths obtained from the mineral samarskite, which had already in 1878 received attention from Delafontaine and later from Lecoq de Boisbaudran. He established the existence of two new elements, samarium and gadolinium, since investigated more especially by Cleve, to whom most of our knowledge on this subject is due. In addition to the rare elements mentioned above, there are a score or so more whose existence is doubtful. Every year is attended by fresh “discoveries” in this prolific source of elementary substances, but the paucity of materials and the predilections of the investigators militate in some measure against a just valuation being accorded to such researches. After having been somewhat neglected for the greater attractions and wider field presented by organic chemistry, the study of the elements and their inorganic compounds is now rapidly coming into favour; new investigators are continually entering the lists; the beaten paths are being retravcrsed and new ramifications pursued.
IV. Organic Chemistry
While inorganic chemistry was primarily developed through the study of minerals—a connexion still shown by the French appellation chimie minérale—organic chemistry owes its origin to the investigation of substances occurring in the vegetable and animal organisms. The quest of the alchemists for the philosopher’s stone, and the almost general adherence of the iatrochemists to the study of the medicinal characters and preparation of metallic compounds, stultified in some measure the investigation of vegetable and animal products. It is true that by the distillation of many herbs, resins and similar substances, several organic compounds had been prepared, and in a few cases employed as medicines; but the prevailing classification of substances by physical and superficial properties led to the correlation of organic and inorganic compounds, without any attention being paid to their chemical composition. The clarification and spirit of research so clearly emphasized by Robert Boyle in the middle of the 17th century is reflected in the classification of substances expounded by Nicolas Lémery, in 1675, in his Cours de chymie. Taking as a basis the nature of the source of compounds, he framed three classes: “mineral,” comprising the metals, minerals, earths and stones; “vegetable,” comprising plants, resins, gums, juices, &c.; and “animal,” comprising animals, their different parts and excreta. Notwithstanding the inconsistency of his allocation of substances to the different groups (for instance, acetic acid was placed in the vegetable class, while the acetates and the products of their dry distillation, acetone, &c., were placed in the mineral class), this classification came into favour. The phlogistonists endeavoured to introduce chemical notions to support it: Becher, in his Physica subterranea(1669), stated that mineral, vegetable and animal matter contained the same elements, but that more simple combinations prevailed in the mineral kingdom; while Stahl, in his Specimen Becherianum (1702), held the “earthy” principle to predominate in the mineral class, and the “aqueous” and “combustible” in the vegetable and animal classes. It thus happened that in the earlier treatises on phlogistic chemistry organic substances were grouped with all combustibles.
The development of organic chemistry from this time until almost the end of the 18th century was almost entirely confined to such compounds as had practical applications, especially in pharmacy and dyeing. A new and energetic spirit was introduced by Scheele; among other discoveries this gifted experimenter isolated and characterized many organic acids, and proved the general occurrence of glycerin (Ölsüss) in all oils and fats. Bergman worked in the same direction; while Rouelle was attracted to the study of animal chemistry. Theoretical speculations were revived by Lavoisier, who, having explained the nature of combustion and determined methods for analysing compounds, concluded that vegetable substances ordinarily contained carbon, hydrogen and oxygen, while animal substances generally contained, in addition to these elements, nitrogen, and sometimes phosphorus and sulphur. Lavoisier, to whom chemistry was primarily the chemistry of oxygen compounds, having developed the radical theory initiated by Guyton de Morveau, formulated the hypothesis that vegetable and animal substances were oxides of radicals composed of carbon and hydrogen; moreover, since simple radicals (the elements) can form more than one oxide, he attributed the same character to his hydrocarbon radicals: he considered, for instance, sugar to be a neutral oxide and oxalic acid a higher oxide of a certain radical, for, when oxidized by nitric acid, sugar yields oxalic acid. At the same time, however, he adhered to the classification of Lémery; and it was only when identical compounds were obtained from both vegetable and animal sources that this subdivision was discarded, and the classes were assimilated in the division organic chemistry.
At this time there existed a belief, held at a later date by Berzelius, Gmelin and many others, that the formation of organic compounds was conditioned by a so-called vital force; and the difficulty of artificially realizing this action explained the supposed impossibility of synthesizing organic compounds. This dogma was shaken by Wöhler’s synthesis of urea in 1828. But the belief died hard; the synthesis of urea remained isolated for many years; and many explanations were attempted by the vitalists (as, for instance, that urea was halfway between the inorganic and organic kingdoms, or that the carbon, from which it was obtained, retained the essentials of this hypothetical vital force), but only to succumb at a later date to the indubitable fact that the same laws of chemical combination prevail in both the animate and inanimate kingdoms, and that the artificial or laboratory synthesis of any substance, either inorganic or organic, is but a question of time, once its constitution is determined.10
The exact delimitation of inorganic and organic chemistry engrossed many minds for many years; and on this point there existed considerable divergence of opinion for several decades. In addition to the vitalistic doctrine of the origin of organic compounds, views based on purely chemical considerations were advanced. The atomic theory, and its correlatives—the laws of constant and multiple proportions—had been shown to possess absolute validity so far as well-characterized inorganic compounds were concerned; but it was open to question whether organic compounds obeyed the same laws. Berzelius, in 1813 and 1814, by improved methods of analysis, established that the Daltonian laws of combination held in both the inorganic and organic kingdoms; and he adopted the view of Lavoisier that organic compounds were oxides of compound radicals, and therefore necessarily contained at least three elements—carbon, hydrogen and oxygen. This view was accepted in 1817 by Leopold Gmelin, who, in his Handbuch der Chemie, regarded inorganic compounds as being of binary composition (the simplest being oxides both acid and basic, which by combination form salts also of binary form), and organic compounds as ternary, i.e. composed of three elements; furthermore, he concluded that inorganic compounds could be synthesized, whereas organic compounds could not. A consequence of this empirical division was that marsh gas, ethylene and cyanogen were regarded as inorganic, and at a later date many other hydrocarbons of undoubtedly organic nature had to be included in the same division.
The binary conception of compounds held by Berzelius received apparent support from the observations of Gay Lussac, in 1815, on the vapour densities of alcohol and ether, which pointed to the conclusion that these substances consisted of one molecule of water and one and two of ethylene respectively; and from Pierre Jean Robiquet and Jean Jacques Colin, showing, in 1816, that ethyl chloride (hydrochloric ether) could be regarded as a compound of ethylene and hydrochloric acid.11 Compound radicals came to be regarded as the immediate constituents of organic compounds; and, at first, a determination of their empirical composition was supposed to be sufficient to characterize them. To this problem there was added another in about the third decade of the 19th century—namely, to determine the manner in which the atoms composing the radical were combined; this supplementary requisite was due to the discovery of the isomerism of silver fulminate and silver cyanate by Justus von Liebig in 1823, and to M. Faraday’s discovery of butylene, isomeric with ethylene, in 1825.
The classical investigation of Liebig and Friedrich Wohler on the radical of benzoic acid (“Über das Radikal der Benzoë-säure,” Ann. Chem., 1832, 3, p. 249) is to be regarded as a most important contribution to the radical theory, for it was shown that a radical containing the elements carbon, hydrogen and oxygen, which they named benzoyl (the termination yl coming from the Gr. ὔλη, matter), formed the basis of benzaldehyde, benzoic acid, benzoyl chloride, benzoyl bromide and benzoyl sulphide, benzamide and benzoic ether. Berzelius immediately appreciated the importance of this discovery, notwithstanding that he was compelled to reject the theory that oxygen could not play any part in a compound radical—a view which he previously considered as axiomatic; and he suggested the names “proin” or “orthrin” (from the Gr. πρωί and ὀρθρός, at dawn). However, in 1833, Berzelius reverted to his earlier opinion that oxygenated radicals were incompatible with his electrochemical theory; he regarded benzoyl as an oxide of the radical C14H10, which he named “picramyl” (from πικρός, bitter, and ἀμυγδάλη, almond), the peroxide being anhydrous benzoic acid; and he dismissed the views of Gay Lussac and Dumas that ethylene was the radical of ether, alcohol and ethyl chloride, setting up in their place the idea that ether was a suboxide of ethyl, (C2H5)2O, which was analogous to K2O, while alcohol was an oxide of a radical C2H6; thus annihilating any relation between these two compounds. This view was modified by Liebig, who regarded ether as ethyl oxide, and alcohol as the hydrate of ethyl oxide; here, however, he was in error, for he attributed to alcohol a molecular weight double its true value. Notwithstanding these errors, the value of the “ethyl theory” was perceived; other radicals—formyl, methyl, amyl, acetyl, &c.—were characterized; Dumas, in 1837, admitted the failure of the etherin theory; and, in company with Liebig, he defined organic chemistry as the “chemistry of compound radicals.” The knowledge of compound radicals received further increment at the hands of Robert W. Bunsen, the discoverer of the cacodyl compounds.
The radical theory, essentially dualistic in nature in view of its similarity to the electrochemical theory of Berzelius, was destined to succumb to a unitary theory. Instances had already been recorded of cases where a halogen element replaced hydrogen with the production of a closely allied substance: Gay Lussac had prepared cyanogen chloride from hydrocyanic acid; Faraday, hexachlorethane from ethylene dichloride, &c. Here the electro-negative halogens exercised a function similar to electro-positive hydrogen. Dumas gave especial attention to such substitutions, named metalepsy μετάληψις, exchange); and framed the following empirical laws to explain the reactions:—(1) a body containing hydrogen when substituted by a halogen loses one atom of hydrogen for every atom of halogen introduced; (2) the same holds if oxygen be present, except that when the oxygen is present as water the latter first loses its hydrogen without replacement, and then substitution according to (1) ensues. Dumas went no further that thus epitomizing his observations; and the next development was made in 1836 by Auguste Laurent, who, having amplified and discussed the applicability of Dumas’ views, promulgated his Nucleus Theory, which assumed the existence of “original nuclei or radicals” (radicaux or noyaux fondamentaux) composed of carbon and hydrogen, and “derived nuclei” (radicaux or noyaux dérivés) formed from the original nuclei by the substitution of hydrogen or the addition of other elements, and having properties closely related to the primary nuclei.
Vigorous opposition was made by Liebig and Berzelius, the latter directing his attack against Dumas, whom he erroneously believed to be the author of what was, in his opinion, a pernicious theory. Dumas repudiated the accusation, affirming that he held exactly contrary views to Laurent; but only to admit their correctness in 1839, when, from his own researches and those of Laurent, Malaguti and Regnault, he formulated his type theory. According to this theory a “chemical type” embraced compounds containing the same number of equivalents combined in a like manner and exhibiting similar properties; thus acetic and trichloracetic acids, aldehyde and chloral, marsh gas and chloroform are pairs of compounds referable to the same type. He also postulated, with Regnault, the existence of “molecular or mechanical types” containing substances which, although having the same number of equivalents, are essentially different in characters. His unitary conceptions may be summarized: every chemical compound forms a complete whole, and cannot therefore consist of two parts; and its chemical character depends primarily upon the arrangement and number of the atoms, and, in a lesser degree, upon their chemical nature. More emphatic opposition to the dualistic theory of Berzelius was hardly possible; this illustrious chemist perceived that the validity of his electrochemical theory was called in question, and therefore he waged vigorous war upon Dumas and his followers. But he fought in a futile cause; to explain the facts put forward by Dumas he had to invent intricate and involved hypotheses, which, it must be said, did not meet with general acceptance; Liebig seceded from him, and invited Wöhler to endeavour to correct him. Still, till the last Berzelius remained faithful to his original theory; experiment, which he had hitherto held to be the only sure method of research, he discarded, and in its place he substituted pure speculation, which greatly injured the radical theory. At the same time, however, the conception of radicals could not be entirely displaced, for the researches of Liebig and Wohler, and those made subsequently by Bunsen, demonstrated beyond all doubt the advantages which would accrue from their correct recognition.
A step forward—the fusion of Dumas’, type theory and the radical theory—was made by Laurent and Charles Gerhardt. As early as 1842, Gerhardt in his Précis de chimie organique exhibited a marked leaning towards Dumas’ theory, and it is without doubt that both Dumas and Laurent exercised considerable influence on his views. Unwilling to discard the strictly unitary views of these chemists, or to adopt the copulae theory of Berzelius, he revived the notion of radicals in a new form. According to Gerhardt, the process of substitution consisted of the union of two residues to form a unitary whole; these residues, previously termed “compound radicals,” are atomic complexes which remain over from the interaction of two compounds. Thus, he interpreted the interaction of benzene and nitric acid as C6H6 + HNO3 = C6H5NO2 + H2O, the “residues” of benzene being C6H5 and H, and of nitric acid HO and NO2. Similarly he represented the reactions investigated by Liebig and Wöhler on benzoyl compounds as double decompositions.
This rejuvenation of the notion of radicals rapidly gained favour; and the complete fusion of the radical theory with the theory of types was not long delayed. In 1849 C.A. Wurtz discovered the amines or substituted ammonias, previously predicted by Liebig; A.W. von Hofmann continued the investigation, and established their recognition as ammonia in which one or more hydrogen atoms had been replaced by hydrocarbon radicals, thus formulating the “ammonia type.” In 1850 A.W. Williamson showed how alcohol and ether were to be regarded as derived from water by substituting one or both hydrogen atoms by the ethyl group; he derived acids and the acid anhydrides from the same type; and from a comparison of many inorganic and the simple organic compounds he concluded that this notion of a “water-type” clarified, in no small measure, the conception of the structure of compounds.
These conclusions were co-ordinated in Gerhardt’s “new theory of types.” Taking as types hydrogen, hydrochloric acid, water and ammonia, he postulated that all organic compounds were referable to these four forms: the hydrogen type included hydrocarbons, aldehydes and ketones; the hydrochloric acid type, the chlorides, bromides and iodides; the water type, the alcohols, ethers, monobasic acids, acid anhydrides, and the analogous sulphur compounds; and the ammonia type, the amines, acid-amides, and the analogous phosphorus and arsenic compounds. The recognition of the polybasicity of acids, which followed from the researches of Thomas Graham and Liebig, had caused Williamson to suggest that dibasic acids could be referred to a double water type, the acid radical replacing an atom of hydrogen in each water molecule; while his discovery of tribasic formic ether, CH(OC2H5)3, in 1854 suggested a triple water type. These views were extended by William Odling, and adopted by Gerhardt, but with modifications of Williamson’s aspects. A further generalization was effected by August Kekulé, who rejected the hydrochloric acid type as unnecessary, and introduced the methane type and condensed mixed types. Pointing out that condensed types can only be fused with a radical replacing more than one atom of hydrogen, he laid the foundation of the doctrine of valency, a doctrine of incalculable service to the knowledge of the structure of chemical compounds.
At about the same time Hermann Kolbe attempted a rehabilitation, with certain modifications, of the dualistic conception of Berzelius. He rejected the Berzelian tenet as to the unalterability of radicals, and admitted that they exercised a considerable influence upon the compounds with which they were copulated. By his own investigations and those of Sir Edward Frankland it was proved that the radical methyl existed in acetic acid; and by the electrolysis of sodium acetate, Kolbe concluded that he had isolated this radical; in this, however, he was wrong, for he really obtained ethane, C2H6, and not methyl, CH3. From similar investigations of valerianic acid he was led to conclude that fatty acids were oxygen compounds of the radicals hydrogen, methyl, ethyl, &c., combined with the double carbon equivalent C2. Thus the radical of acetic acid, acetyl,12 was C2H3·C2. (It will be noticed that Kolbe used the atomic weights H=1, C=6, O=8, S=16, &c.; his formulae, however, were molecular formulae, i.e. the molecular weights were the same as in use to-day.) This connecting link, C2, was regarded as essential, while the methyl, ethyl, &c. was but a sort of appendage; but Kolbe could not clearly conceive the manner of copulation.
The brilliant researches of Frankland on the organo-metallic compounds, and his consequent doctrine of saturation capacity or valency of elements and radicals, relieved Kolbe’s views of all obscurity. The doctrine of copulae was discarded, and in 1859 emphasis was given to the view that all organic compounds were derivatives of inorganic by simple substitution processes. He was thus enabled to predict compounds then unknown, e.g. the secondary and tertiary alcohols; and with inestimable perspicacity he proved intimate relations between compounds previously held to be quite distinct. Lactic acid and alanine were shown to be oxy- and amino-propionic acids respectively; glycollic acid and glycocoll, oxy- and amino-acetic acids; salicylic and benzamic acids, oxy- and amino-benzoic acids.
Another consequence of the doctrine of valency was that it permitted the graphic representation of the molecule. The “structure theory” (or the mode of linking of the atoms) of carbon compounds, founded by Butlerow, Kekulé and Couper and, at a later date, marvellously enhanced by the doctrine of stereo-isomerism, due to J.H. van’t Hoff and Le Bel, occupies such a position in organic chemistry that its value can never be transcended. By its aid the molecule is represented as a collection of atoms connected together by valencies in such a manner that the part played by each atom is represented; isomerism, or the existence of two or more chemically different substances having identical molecular weights, is adequately shown; and, most important of all, once the structure is determined, the synthesis of the compound is but a matter of time.
In this summary the leading factors which have contributed to a correct appreciation of organic compounds have so far been considered historically, but instead of continuing this method it has been thought advisable to present an epitome of present-day conclusions, not chronologically, but as exhibiting the principles and subject-matter of our science.
Classification of Organic Compounds.
An apt definition of organic chemistry is that it is “the study of the hydrocarbons and their derivatives.” This description, although not absolutely comprehensive, serves as a convenient starting-point for a preliminary classification, since a great number of substances, including the most important, are directly referable to hydrocarbons, being formed by replacing one or more hydrogen atoms by other atoms or groups. Two distinct types of hydrocarbons exist: (1) those consisting of an open chain of carbon atoms—named the “aliphatic series” (ἄλειφαρ, oil or fat), and (2) those consisting of a closed chain—the “carbocyclic series.” The second series can be further divided into two groups: (1) those exhibiting properties closely analogous to the aliphatic series—the polymethylenes (q.v.), and (2) a series exhibiting properties differing in many respects from the aliphatic and polymethylene compounds, and characterized by a peculiar stability which is to be associated with the disposition of certain carbon valencies not saturated by hydrogen—the “aromatic series.” There also exists an extensive class of compounds termed the “heterocyclic series”—these compounds are derived from ring systems containing atoms other than carbon; this class is more generally allied to the aromatic series than to the aliphatic.
We now proceed to discuss the types of aliphatic compounds; then, the characteristic groupings having been established, an epitome of their derivatives will be given. Carbocyclic rings will next be treated, benzene and its allies in some detail; and finally the heterocyclic nuclei.
Accepting the doctrine of the tetravalency of carbon (its divalency in such compounds as carbon monoxide, various isocyanides, fulminic acid, &c., and its possible trivalency in M. Gomberg’s triphenyl-methyl play no part in what follows), it is readily seen that the simplest hydrocarbon has the formula CH4 named methane, in which the hydrogen atoms are of equal value, and which may be pictured as placed at the vertices of a tetrahedron, the carbon atom occupying the centre. This tetrahedral configuration is based on the existence of only one methylene dichloride, two being necessary if the carbon valencies were directed from the centre of a plane square to its corners, and on the existence of two optical isomers of the formula C.A.B.D.E., C being a carbon atom and A.B.D.E. being different monovalent atoms or radicals (see Stereo-Isomerism). The equivalence of the four hydrogen atoms of methane rested on indirect evidence, e.g. the existence of only one acetic acid, methyl chloride, and other monosubstitution derivatives—until the experimental proof by L. Henry (Zeit. f. Phys. Chem., 1888, 2, p. 553), who prepared the four nitromethanes, CH3NO2, each atom in methane being successively replaced by the nitro-group.
Henry started with methyl iodide, the formula of which we write in the form CIaHbHcHd. This readily gave with silver nitrite a nitromethane in which we may suppose the nitro-group to replace the a iodine atom, i.e. C(NO2)aHbHcHd. The same methyl iodide gave with potassium cyanide, acetonitril, which was hydrolysed to acetic acid; this must be C(COOH)aHbHcHd. Chlorination of this substance gave a monochloracetic acid; we will assume the chlorine atom to replace the b hydrogen atom. This acid with silver nitrite gave nitroacetic acid, which readily gave the second nitromethane, CHa(NO2)bHcHd identical with the first nitromethane. From the nitroacetic acid obtained above, malonic acid was prepared, and from this a monochlormalonic acid was obtained; we assume the chlorine atom to replace the c hydrogen atom. This acid gives with silver nitrite the corresponding nitromalonic acid, which readily yielded the third nitromethane, CHaHb(NO2)cHd, also identical with the first. The fourth nitromethane was obtained from the nitromalonic acid previously mentioned by a repetition of the method by which the third was prepared; this was identical with the other three.
Let us now consider hydrocarbons containing 2 atoms of carbon. Three such compounds are possible according to the number of valencies acting directly between the carbon atoms. Thus, if they are connected by one valency, and the remaining valencies saturated by hydrogen, we obtain the compound H3C·CH3, ethane. This compound may be considered as derived from methane, CH4, by replacing a hydrogen atom by the monovalent group CH3, known as methyl; hence ethane may be named “methylmethane.” If the carbon atoms are connected by two valencies, we obtain a compound H2C:CH2, ethylene; if by three valencies, HC∶CH, acetylene. These last two compounds are termed unsaturated, whereas ethane is saturated. It is obvious that we have derived three combinations of carbon with hydrogen, characterized by containing a single, double, and triple linkage; and from each of these, by the substitution of a methyl group for a hydrogen atom, compounds of the same nature result. Thus ethane gives H3C·CH2·CH3, propane; ethylene gives H2C:CH·CH3, propylene; and acetylene gives HC∶C·CH3, allylene. By continuing the introduction of methyl groups we obtain three series of homologous hydro-carbons given, by the general formulae CnH2n+2, CnH2n, and CnH2n-2, each member differing from the preceding one of the same series by CH2. It will be noticed that compounds containing two double linkages will have the same general formula as the acetylene series; such compounds are known as the “diolefines.” Hydrocarbons containing any number of double or triple linkages, as well as both double and triple linkages, are possible, and a considerable number of such compounds have been prepared.
A more complete idea of the notion of a compound radical follows from a consideration of the compound propane. We derived this substance from ethane by introducing a methyl group; hence it may be termed “methylethane.” Equally well we may derive it from methane by replacing a hydrogen atom by the monovalent group CH2·CH3, named ethyl; hence propane may be considered as “ethylmethane.” Further, since methane may be regarded as formed by the conjunction of a methyl group with a hydrogen atom, it may be named “methyl hydride”; similarly ethane is “ethyl hydride,” propane, “propyl hydride,” and so on. The importance of such groups as methyl, ethyl, &c. in attempting a nomenclature of organic compounds cannot be overestimated; these compound radicals, frequently termed alkyl radicals, serve a similar purpose to the organic chemist as the elements to the inorganic chemist.
In methane and ethane the hydrogen atoms are of equal value, and no matter which one may be substituted by another element or group the same compound will result. In propane, on the other hand, the hydrogen atoms attached to the terminal carbon atoms differ from those joined to the medial atom; we may therefore expect to obtain different compounds according to the position of the hydrogen atom substituted. By introducing a methyl group we may obtain CH3·CH2·CH2·CH3, known as “normal” or n-butane, substitution occurring at a terminal atom, or CH3·CH(CH3)·CH3, isobutane, substitution occurring at the medial atom. From n-butane we may derive, by a similar substitution of methyl groups, the two hydrocarbons: (1) CH3·CH2·CH2·CH2·CH3, and (2) CH3·CH(CH3)·CH2·CH3; from isobutane we may also derive two compounds, one identical with (2.), and a new one (3) CH3(CH3)C(CH3)CH3. These three hydrocarbons are isomeric, i.e. they possess the same formula, but differ in constitution. We notice that they may be differentiated as follows: (1) is built up solely of methyl and ·CH2· (methylene) groups and the molecule consists of a single chain; such hydrocarbons are referred to as being normal; (2) has a branch and contains the group ÷CH (methine) in which the free valencies are attached to carbon atoms; such hydrocarbons are termed secondary or iso-; (3) is characterized by a carbon atom linked directly to four other carbon atoms; such hydrocarbons are known as tertiary.
Deferring the detailed discussion of cyclic or ringed hydrocarbons, a correlation of the various types or classes of compounds which may be derived from hydrocarbon nuclei will now be given. It will be seen that each type depends upon a specific radical or atom, and the copulation of this character with any hydrocarbon radical (open or cyclic) gives origin to a compound of the same class.
It is convenient first to consider the effect of introducing one, two, or three hydroxyl (OH) groups into the -CH3, >CH2, and ->CH groups, which we have seen to characterize the different types of hydrocarbons. It may be noticed here that cyclic nuclei can only contain the groups >CH2 and ->CH, the first characterizing the polymethylene and reduced heterocyclic compounds, the second true aromatic compounds.
Substituting one hydroxyl group into each of these residues, we obtain radicals of the type -CH2·OH, >CH·OH, and ->C·OH; these compounds are known as alcohols (q.v.), and are termed primary, secondary, and tertiary respectively. Polymethylenes can give only secondary and tertiary alcohols, benzene only tertiary; these latter compounds are known as phenols. A second hydroxyl group may be introduced into the residues -CH2·OH and >CH·OH, with the production of radicals of the form -CH(OH)2 and >C(OH)2. Compounds containing these groupings are, however, rarely observed (see Chloral), and it is generally found that when compounds of these types are expected, the elements of water are split off, and the typical groupings are reduced to -CH:O and >C:O. Compounds containing the group -CH:O are known as aldehydes (q.v.), while the group >C:O (sometimes termed the carbonyl or keto group) characterizes the ketones (q.v.). A third hydroxyl group may be introduced into the -CH:O residue with the formation of the radical -C(OH):O; this is known as the carboxyl group, and characterizes the organic acids.
Sulphur analogues of these oxygen compounds are known. Thus the thio-alcohols or mercaptans (q.v.) contain the group -CH2·SH; and the elimination of the elements of sulphuretted hydrogen between two molecules of a thio-alcohol results in the formation of a thio-ether or sulphide, R2S. Oxidation of thio-ethers results in the formation of sulphoxides, R2:S:O, and sulphones, R2:SO2; oxidation of mercaptans yields sulphonic acids, R·SO3H, and of sodium mercaptides sulphinic acids, R·SO(OH). We may also notice that thio-ethers combine with alkyl iodides to form sulphine or sulphonium compounds, R3÷SI. Thio-aldehydes, thio-ketones and thio-acids also exist.
We proceed to consider various simple derivatives of the alcohols, which we may here regard as hydroxy hydrocarbons, R·OH, where R is an alkyl radical, either aliphatic or cyclic in nature.
Of these, undoubtedly the simplest are the ethers (q.v.), formed by the elimination of the elements of water between two molecules of the same alcohol, “simple ethers,” or of different alcohols, “mixed ethers.” These compounds may be regarded as oxides in just the same way as the alcohols are regarded as hydroxides. In fact, the analogy between the alkyl groups and metallic elements forms a convenient basis from which to consider many derivatives. Thus from ethyl alcohol there can be prepared compounds, termed esters (q.v.), or ethereal salts, exactly comparable in structure with corresponding salts of, say, potassium; by the action of the phosphorus haloids, the hydroxyl group is replaced by a halogen atom with the formation of derivatives of the type R·Cl(Br,I); nitric acid forms nitrates, R·O·NO2; nitrous acid, nitrites, R·O·NO; sulphuric acid gives normal sulphates R2SO4, or acid sulphates, R·SO4H. Organic acids also condense with alcohols to form similar compounds: the fats, waxes, and essential oils are naturally occurring substances of this class.
An important class of compounds, termed amines (q.v.), results from the condensation of alcohols with ammonia, water being eliminated between the alcoholic hydroxyl group and a hydrogen atom of the ammonia. Three types of amines are possible and have been prepared: primary, R·NH2; secondary, R2:NH; and tertiary, R3÷N; the oxamines, R3N:O, are closely related to the tertiary ammonias, which also unite with a molecule of alkyl iodide to form salts of quaternary ammonium bases, e.g. R4N·I. It is worthy of note that phosphorus and arsenic bases analogous to the amines are known (see Phosphorus and Arsenic). From the primary amines are derived the diazo compounds (q.v.) and azo compounds (q.v.); closely related are the hydrazines (q.v.). Secondary amines yield nitrosamines, R2N·NO, with nitrous acid. By the action of hydroxylamine or phenylhydrazine on aldehydes or ketones, condensation occurs between the carbonyl oxygen of the aldehyde or ketone and the amino group of the hydroxylamine or hydrazine. Thus with hydroxylamine aldehydes yield aldoximes, R·CH:N·OH, and ketones, ketoximes, R2C:N·OH (see Oximes), while phenyl hydrazine gives phenylhydrazones, R2C:N·NH·C6H5 (see Hydrazones). Oxyaldehydes and oxyketones (viz. compounds containing an oxy in addition to an aldehydic or ketonic group) undergo both condensation and oxidation when treated with phenylhydrazine, forming compounds known as osozones; these are of great importance in characterizing the sugars (q.v.).
The carboxyl group constitutes another convenient starting-point for the orientation of many types of organic compounds. This group may be considered as resulting from the fusion of a carbonyl (:CO) and a hydroxyl (HO·) group; and we may expect to meet with compounds bearing structural resemblances to the derivatives of alcohols and aldehydes (or ketones).
Considering derivatives primarily concerned with transformations of the hydroxyl group, we may regard our typical acid as a fusion of a radical R·CO- (named acetyl, propionyl, butyl, &c., generally according to the name of the hydrocarbon containing the same number of carbon atoms) and a hydroxyl group. By replacing the hydroxyl group by a halogen, acid-haloids result; by the elimination of the elements of water between two molecules, acid-anhydrides, which may be oxidized to acid-peroxides; by replacing the hydroxyl group by the group ·SH, thio-acids; by replacing it by the amino group, acid-amides (q.v.); by replacing it by the group -NH·NH2, acid-hydrazides. The structural relations of these compounds are here shown:
| R·CO·OH; | R·CO·Cl; | (R·CO)2O; | R·CO·SH; |
| acid; | acid-chloride; | acid-anhydride; | thio-acid; |
| R·CO·NH2; | R·CO·NH·NH2. |
| acid-amide; | acid-hydrazide. |
It is necessary clearly to distinguish such compounds as the amino- (or amido-) acids and acid-amides; in the first case the amino group is substituted in the hydrocarbon residue, in the second it is substituted in the carboxyl group.
By transformations of the carbonyl group, and at the same time of the hydroxyl group, many interesting types of nitrogen compounds may be correlated.
Thus from the acid-amides, which we have seen to be closely related to the acids themselves, we obtain, by replacing the carbonyl oxygen by chlorine, the acidamido-chlorides, R·CCl2·NH2, from which are derived the imido-chlorides, R·CCl:NH, by loss of one molecule of hydrochloric acid. By replacing the chlorine in the imido-chloride by an oxyalkyl group we obtain the imido-ethers, R·C(OR’):NH; and by an amino group, the amidines, R·C(NH2):NH. The carbonyl oxygen may also be replaced by the oxime group, :N·OH; thus the acids yield the hydroxamic acids, R·C(OH):NOH, and the acid-amides the amidoximes, R·C(NH2):NOH. Closely related to the amidoximes are the nitrolic acids, R·C(NO2):NOH.
Cyclic Hydrocarbons and Nuclei.
Having passed in rapid review the various types of compounds derived by substituting for hydrogen various atoms or groups of atoms in hydrocarbons (the separate articles on specific compounds should be consulted for more detailed accounts), we now proceed to consider the closed chain compounds. Here we meet with a great diversity of types: oxygen, nitrogen, sulphur and other elements may, in addition to carbon, combine together in a great number of arrangements to form cyclic nuclei, which exhibit characters closely resembling open-chain compounds in so far as they yield substitution derivatives, and behave as compound radicals. In classifying closed chain compounds, the first step consists in dividing them into: (1) carbocyclic, in which the ring is composed solely of carbon atoms—these are also known as homocyclic or isocyclic on account of the identity of the members of the ring—and (2) heterocyclic, in which different elements go to make up the ring. Two primary divisions of carbocyclic compounds may be conveniently made: (1) those in which the carbon atoms are completely saturated—these are known by the generic term polymethylenes, their general formula being (CH2)n: it will be noticed that they are isomeric with ethylene and its homologues; they differ, however, from this series in not containing a double linkage, but have a ringed structure; and (2) those containing fewer hydrogen atoms than suffice to saturate the carbon valencies—these are known as the aromatic compounds proper, or as benzene compounds, from the predominant part which benzene plays in their constitution.
It was long supposed that the simplest ring obtainable contained six atoms of carbon, and the discovery of trimethylene in 1882 by August Freund by the action of sodium on trimethylene bromide, Br(CH2)3Br, came somewhat as a surprise, especially in view of its behaviour with bromine and hydrogen bromide. In comparison with the isomeric propylene, CH3·HC:CH2, it is remarkably inert, being only very slowly attacked by bromine, which readily combines with propylene. But on the other hand, it is readily converted by hydrobromic acid into normal propyl bromide, CH3·CH2·CH2Br. The separation of carbon atoms united by single affinities in this manner at the time the observation was made was altogether without precedent. A similar behaviour has since been noticed in other trimethylene derivatives, but the fact that bromine, which usually acts so much more readily than hydrobromic acid on unsaturated compounds, should be so inert when hydrobromic acid acts readily is one still needing a satisfactory explanation. A great impetus was given to the study of polymethylene derivatives by the important and unexpected observation made by W.H. Perkin, junr., in 1883, that ethylene and trimethylene bromides are capable of acting in such a way on sodium acetoacetic ester as to form tri- and tetra-methylene rings. Perkin has himself contributed largely to our knowledge of such compounds; penta- and hexa-methylene derivatives have also received considerable attention (see Polymethylenes).
A. von Baeyer has sought to explain the variations in stability manifest in the various polymethylene rings by a purely mechanical hypothesis, the “strain” or Spannungs theory (Ber., 1885, p. 2277). Assuming the four valencies of the carbon atom to be directed from the centre of a regular tetrahedron towards its four corners, the angle at which they meet is 109° 28′. Baeyer supposes that in the formation of carbon ~52 “rings” the valencies become deflected from their positions, and that the tension thus introduced may be deduced from a comparison of this angle with the angles at which the strained valencies would meet. He regards the amount of deflection as a measure of the stability of the “ring.” The readiness with which ethylene is acted on in comparison with other types of hydrocarbon, for example, is in harmony, he considers, with the circumstance that the greatest distortion must be involved in its formation, as if deflected into parallelism each valency will be drawn out of its position through ½.109° 28′. The values in other cases are calculable from the formula ½(1O9° 28′ - a), where a is the internal angle of the regular polygon contained by sides equal in number to the number of the carbon atoms composing the ring. These values are:—
| Trimethylene. | Tetramethylene. |
| ½(109° 28′ - 60°) = 24° 44′. | ½(109° 28′ - 90°) = 9° 44′. |
| Pentamethylene. | Hexamethylene. |
| ½(109° 28′ - 108°) = 0° 44′. | ½(109° 28′ - 120°) = -5° 16′. |
The general behaviour of the several types of hydrocarbons is certainly in accordance with this conception, and it is a remarkable fact that when benzene is reduced with hydriodic acid, it is converted into a mixture of hexamethylene and methylpentamethylene (cf. W. Markownikov, Ann., 1898, 302, p. 1); and many other cases of the conversion of six-carbon rings into five-carbon rings have been recorded (see below, Decompositions of the Benzene Ring). Similar considerations will apply to rings containing other elements besides carbon. As an illustration it may be pointed out that in the case of the two known types of lactones—the γ-lactones, which contain four carbon atoms and one oxygen atom in the ring, are more readily formed and more stable (less readily hydrolysed) than the δ-lactones, which contain one oxygen and five carbon atoms in the ring. That the number of atoms which can be associated in a ring by single affinities is limited there can be no doubt, but there is not yet sufficient evidence to show where the limit must be placed. Baeyer has suggested that his hypothesis may also be applied to explain the instability of acetylene and its derivatives, and the still greater instability of the polyacetylene compounds.
Benzene.
The ringed structure of benzene, C6H6, was first suggested in 1865 by August Kekulé, who represented the molecule by six CH groups placed at the six angles of a regular hexagon, the sides of which denoted the valencies saturated by adjacent carbon atoms, the fourth valencies of each carbon atom being represented as saturated along alternate sides. This formula, notwithstanding many attempts at both disproving and modifying it, has well stood the test of time; the subject has been the basis of constant discussion, many variations have been proposed, but the original conception of Kekulé remains quite as convenient as any of the newer forms, especially when considering the syntheses and decompositions of the benzene complex. It will be seen, however, that the absolute disposition of the fourth valency may be ignored in a great many cases, and consequently the complex may be adequately represented as a hexagon. This symbol is in general use; it is assumed that at each corner there is a CH group which, however, is not always written in; if a hydrogen atom be substituted by another group, then this group is attached to the corner previously occupied by the displaced hydrogen. The following diagrams illustrate these statements:—
Benzene. Abbreviated. Oxybenzene. Abbreviated.
From the benzene nucleus we can derive other aromatic nuclei, graphically represented by fusing two or more hexagons along common sides. By fusing two nuclei we obtain the formula of naphthalene, C10H10; by fusing three, the hydrocarbons anthracene and phenanthrene, C14H10; by fusing four, chrysene, C18H12, and possibly pyrene, C16H10; by fusing five, picene, C22H14. But it must be here understood that each member of these condensed nuclei need not necessarily be identical in structure; thus the central nuclei in anthracene and phenanthrene differ very considerably from the terminal nuclei (see below, Condensed Nuclei). Other hydrocarbon nuclei generally classed as aromatic in character result from the union of two or more benzene nuclei joined by one or two valencies with polymethylene or oxidized polymethylene rings; instances of such nuclei are indene, hydrindene, fluorene, and fluor-anthene. From these nuclei an immense number of derivatives may be obtained, for the hydrogen atoms may be substituted by any of the radicals discussed in the preceding section on the classification of organic compounds.
We now proceed to consider the properties, syntheses, decompositions and constitution of the benzene complex. It has already been stated that benzene derivatives may be Distinctions between aliphatic and aromatic compounds. regarded as formed by the replacement of hydrogen atoms by other elements or radicals in exactly the same manner as in the aliphatic series. Important differences, however, are immediately met with when we consider the methods by which derivatives are obtained. For example: nitric acid and sulphuric acid readily react with benzene and its homologues with the production of nitro derivatives and sulphonic acids, while in the aliphatic series these acids exert no substituting action (in the case of the olefines, the latter acid forms an addition product); another distinction is that the benzene complex is more stable towards oxidizing agents. This and other facts connected with the stability of benzenoid compounds are clearly shown when we consider mixed aliphatic-aromatic hydrocarbons, i.e. compounds derived by substituting aliphatic radicals in the benzene nucleus; such a compound is methylbenzene or toluene, C6H5·CH3. This compound is readily oxidized to benzoic acid, C6H5·COOH, the aromatic residue being unattacked; nitric and sulphuric acids produce nitro-toluenes, C6H4·CH3·NO2, and toluene sulphonic acids, C6H4·CH3·SO3H; chlorination may result in the formation of derivatives substituted either in the aromatic nucleus or in the side chain; the former substitution occurs most readily, chlor-toluenes, C6H4·CH3·Cl, being formed, while the latter, which needs an elevation in temperature or other auxiliary, yields benzyl chloride, C6H5·CH2Cl, and benzal chloride, C6H5·CHCl2. In general, the aliphatic residues in such mixed compounds retain the characters of their class, while the aromatic residues retain the properties of benzene.
Further differences become apparent when various typical compounds are compared. The introduction of hydroxyl groups into the benzene nucleus gives rise to compounds generically named phenols, which, although resembling the aliphatic alcohols in their origin, differ from these substances in their increased chemical activity and acid nature. The phenols more closely resemble the tertiary alcohols, since the hydroxyl group is linked to a carbon atom which is united to other carbon atoms by its remaining three valencies; hence on oxidation they cannot yield the corresponding aldehydes, ketones or acids (see below, Decompositions of the Benzene Ring). The amines also exhibit striking differences: in the aliphatic series these compounds may be directly formed from the alkyl haloids and ammonia, but in the benzene series this reaction is quite impossible unless the haloid atom be weakened by the presence of other substituents, e.g. nitro groups. Moreover, while methylamine, dimethylamine, and trimethylamine increase in basicity corresponding to the introduction of successive methyl groups, phenylamine or aniline, diphenylamine, and triphenylamine are in decreasing order of basicity, the salts of diphenylamine being decomposed by water. Mixed aromatic-aliphatic amines, both secondary and tertiary, are also more strongly basic than the pure aromatic amines, and less basic than the true aliphatic compounds; e.g. aniline, C6H5·NH2, monomethyl aniline, C6H5·NH·CH3, and dimethyl aniline, C6H5·N(CH3)2, are in increasing order of basicity. These observations may be summarized by saying that the benzene nucleus is more negative in character than the aliphatic residues.
Isomerism of Benzene Derivatives.—Although Kekulé founded his famous benzene formula in 1865 on the assumptions that the six hydrogen atoms in benzene are equivalent and that the molecule is symmetrical, i.e. that two pairs of hydrogen atoms are symmetrically situated with reference to any specified hydrogen atom, the absolute demonstration of the validity of these assumptions was first given by A. Ladenburg in 1874 (see Ber., 1874, 7, p. 1684; 1875, 8, p. 1666; Theorie der aromatischen Verbindungen, 1876). These results may be graphically represented as follows: numbering the hydrogen atoms in cyclical order from 1 to 6, then the first thesis demands that whichever atom is substituted the same compound results, while the second thesis points out that the pairs 2 and 6, and 3 and 5 are symmetrical with respect to 1, or in other words, the di-substitution derivatives 1.2 and 1.6, and also 1.3 and 1.5 are identical. Therefore three di-derivatives are possible, viz. 1.2 or 1.6, named ortho- (o), 1.3 or 1.5, named meta- (m), and 1.4, named para- compounds (p). In the same way it may be shown that three tri-substitution, three tetra-substitution, one penta-substitution, and one hexa-substitution derivative are possible. Of the tri-substitution derivatives, 1.2.3.-compounds are known as “adjacent” or “vicinal” (v), the 1.2.4 as “asymmetrical” (as), the 1.3.5 as “symmetrical” (s); of the tetra-substitution derivatives, 1.2.3.4-compounds are known as “adjacent,” 1.2.3.5 as “asymmetrical,” and 1.2.4.5 as “symmetrical.”
Here we have assumed the substituent groups to be alike; when they are unlike, a greater number of isomers is possible. Thus in the tri-substitution derivatives six isomers, and no more, are possible when two of the substituents are alike; for instance, six diaminobenzoic acids, C6H3(NH2)2COOH, are known; when all are unlike ten isomers are possible; thus, ten oxytoluic acids, C6H3·CH3·OH·COOH, are known. In the case of tetra-substituted compounds, thirty isomers are possible when all the groups are different.
The preceding considerations render it comparatively easy to follow the reasoning on which the experimental verification of the above statements is based. The proof is divided into two Equivalence of four hydrogen atoms. parts: (1) that four hydrogen atoms are equal, and (2) that two pairs of hydrogen atoms are symmetrical with reference to a specified hydrogen atom. In the first thesis, phenol or oxybenzene, C6H5·OH, in which we will assume the hydroxyl group to occupy position 1, is converted into brombenzene, which is then converted into benzoic acid, C6H5·COOH. From this substance, an oxybenzoic acid (meta-), C6H4·OH·COOH, may be prepared; and the two other known oxybenzoic acids (ortho- and para-) may be converted into benzoic acid. These three acids yield on heating phenol, identical with the substance started with, and since in the three oxybenzoic acids the hydroxyl groups must occupy positions other than 1, it follows that four hydrogen atoms are equal in value.
R. Hübner and A. Petermann (Ann., 1869, 149, p. 129) provided the proof of the equivalence of the atoms 2 and 6 with respect to 1. From meta-brombenzoic acid two nitrobrombenzoic Symmetry of pairs of hydrogen atoms. acids are obtained on direct nitration; elimination of the bromine atom and the reduction of the nitro to an amino group in these two acids results in the formation of the same ortho-aminobenzoic acid. Hence the positions occupied by the nitro groups in the two different nitrobrombenzoic acids must be symmetrical with respect to the carboxyl group. In 1879, Hübner (Ann., 195, p. 4) proved the equivalence of the second pair, viz. 3 and 5, by starting out with ortho-aminobenzoic acid, previously obtained by two different methods. This substance readily yields ortho-oxybenzoic acid or salicylic acid, which on nitration yields two mononitro-oxybenzoic acids. By eliminating the hydroxy groups in these acids the same nitrobenzoic acid is obtained, which yields on reduction an aminobenzoic acid different from the starting-out acid. Therefore there must be another pair of hydrogen atoms, other than 2 and 6, which are symmetrical with respect to 1. The symmetry of the second pair was also established in 1878 by E. Wroblewsky (Ann., 192, p. 196).
Orientation of Substituent Groups.—The determination of the relative positions of the substituents in a benzene derivative constitutes an important factor in the general investigation of such compounds. Confining our attention, for the present, to di-substitution products we see that there are three distinct series of compounds to be considered. Generally if any group be replaced by another group, then the second group enters the nucleus in the position occupied by the displaced group; this means that if we can definitely orientate three di-derivatives of benzene, then any other compound, which can be obtained from or converted into one of our typical derivatives, may be definitely orientated. Intermolecular transformations—migrations of substituent groups from one carbon atom to another—are of fairly common occurrence among oxy compounds at elevated temperatures. Thus potassium ortho-oxybenzoate is converted into the salt of para-oxybenzoic acid at 220°; the three bromphenols, and also the brombenzenesulphonic acids, yield m-dioxybenzene or resorcin when fused with potash. It is necessary, therefore, to avoid reactions involving such intermolecular migrations when determining the orientation of aromatic compounds.
Such a series of typical compounds are the benzene dicarboxylic acids (phthalic acids), C6H4(COOH)2. C. Graebe (Ann., 1869, 149, p. 22) orientated the ortho-compound or phthalic acid from its formation from naphthalene on oxidation; the meta-compound or isophthalic acid is orientated by its production from mesitylene, shown by A. Ladenburg (Ann., 1875, 179, p. 163) to be symmetrical trimethyl benzene; terephthalic acid, the remaining isomer, must therefore be the para-compound.
P. Griess (Ber., 1872, 5, p. 192; 1874, 7, p. 1223) orientated the three diaminobenzenes or phenylene diamines by considering their preparation by the elimination of the carboxyl group in the six diaminobenzoic acids. The diaminobenzene resulting from two of these acids is the ortho-compound; from three, the meta-; and from one the para-; this is explained by the following scheme:—
W. Körner (Gazz. Chem. Ital., 4, p. 305) in 1874 orientated the three dibrombenzenes in a somewhat similar manner. Starting with the three isomeric compounds, he found that one gave two tribrombenzenes, another gave three, while the third gave only one. A scheme such as the preceding one shows that the first dibrombenzene must be the ortho-compound, the second the meta-, and the third the para-derivative. Further research in this direction was made by D.E. Noetling (Ber., 1885, 18, p. 2657), who investigated the nitro-, amino-, and oxy-xylenes in their relations to the three xylenes or dimethyl benzenes.
The orientation of higher substitution derivatives is determined by considering the di- and tri-substitution compounds into which they can be transformed.
Substitution of the Benzene Ring.—As a general rule, homologues and mono-derivatives of benzene react more readily with substituting agents than the parent hydrocarbon; for example, phenol is converted into tribromphenol by the action of bromine water, and into the nitrophenols by dilute nitric acid; similar activity characterizes aniline. Not only does the substituent group modify the readiness with which the derivative is attacked, but also the nature of the product. Starting with a mono-derivative, we have seen that a substituent group may enter in either of three positions to form an ortho-, meta-, or para-compound. Experience has shown that such mono-derivatives as nitro compounds, sulphonic acids, carboxylic acids, aldehydes, and ketones yield as a general rule chiefly the meta-compounds, and this is independent of the nature of the second group introduced; on the other hand, benzene haloids, amino-, homologous-, and hydroxy-benzenes yield principally a mixture of the ortho- and para-compounds. These facts are embodied in the “Rule of Crum Brown and J. Gibson” (Jour. Chem. Soc. 61, p. 367): If the hydrogen compound of the substituent already in the benzene nucleus can be directly oxidized to the corresponding hydroxyl compound, then meta-derivatives predominate on further substitution, if not, then ortho- and para-derivatives. By further substitution of ortho- and para-di-derivatives, in general the same tri-derivative [1.2.4] is formed (Ann., 1878, 192, p. 219); meta-compounds yield [1.3.4] and [1.2.3] tri-derivatives, except in such cases as when both substituent groups are strongly acid, e.g. m-dinitrobenzene, then [1.3.5]-derivatives are obtained.
Syntheses of the Benzene Ring.—The characteristic distinctions which exist between aliphatic and benzenoid compounds make the transformations of one class into the other especially interesting.
In the first place we may notice a tendency of several aliphatic compounds, e.g. methane, tetrachlormethane, &c., to yield aromatic compounds when subjected to a high temperature, the so-called pyrogenetic reactions (from Greek πῦρ, fire, and γεννάω, I produce); the predominance of benzenoid, and related compounds—naphthalene, anthracene, phenanthrene, &c.—in coal-tar is probably to be associated with similar pyrocondensations. Long-continued treatment with halogens may, in some cases, result in the formation of aromatic compounds; thus perchlorbenzene, C6Cl6, frequently appears as a product of exhaustive chlorination, while hexyl iodide, C6H13I, yields perchlor- and perbrom-benzene quite readily.
The trimolecular polymerization of numerous acetylene compounds—substances containing two trebly linked carbon atoms, —C:C—, to form derivatives of benzene is of considerable interest. M.P.E. Berthelot first accomplished the synthesis of benzene in 1870 by leading acetylene, HC÷CH, through tubes heated to dull redness; at higher temperatures the action becomes reversible, the benzene yielding diphenyl, diphenylbenzene, and acetylene. The condensation of acetylene to benzene is also possible at ordinary temperatures by leading the gas over pyrophoric iron, nickel, cobalt, or spongy platinum (P. Sabatier and J.B. Senderens). The homologues of acetylene condense more readily; thus allylene, CH÷C·CH3, and crotonylene, CH3·C∶C·CH3, yield trimethyl- and hexamethyl-benzene under the influence of sulphuric acid. Toluene or mono-methylbenzene results from the pyrocondensation of a mixture of acetylene and allylene. Substituted acetylenes also exhibit this form of condensation; for instance, bromacetylene, BrC∶CH, is readily converted into tribrombenzene, while propiolic acid, HC∶C·COOH, under the influence of sunlight, gives benzene tricarboxylic acid.
A larger and more important series of condensations may be grouped together as resulting from the elimination of the elements of water between carbonyl (CO) and methylene (CH2) groups. A historic example is that of the condensation of three molecules of acetone, CH3·CO·CH3, in the presence of sulphuric acid, to s-trimethylbenzene or mesitylene, C6H3(CH3)3, first observed in 1837 by R. Kane; methylethyl ketone and methyl-n-propyl ketone suffer similar condensations to s-triethylbenzene and s-tri-n-propylbenzene respectively. Somewhat similar condensations are: of geranial or citral, (CH3)2CH·CH2·CH:CH·C(CH3):CH·CHO, to p-isopropyl-methylbenzene or cymene; of the condensation product of methylethylacrolein and acetone, CH3·CH2·CH:C(CH3)·CH:CH·CO·CH3, to [1.3.4]-trimethylbenzene or pseudocumene; and of the condensation product of two molecules of isovaleryl aldehyde with one of acetone, C3H7·CH2·CH:C(C3H7)·CH:CH·CO·CH3, to (1)-methyl-2-4-di-isopropyl benzene. An analogous synthesis is that of di-hydro-m-xylene from methyl heptenone, (CH3)2C:CH·(CH2)2·CO·CH3. Certain a-diketones condense to form benzenoid quinones, two molecules of the diketone taking part in the reaction; thus diacetyl, CH3·CO·CO·CH3, yields p-xyloquinone, C6H2(CH3)2O2 (Ber., 1888, 21, p. 1411), and acetylpropionyl, CH3·CO·CO·C2H5, yields duroquinone, or tetramethylquinone, C6(CH3)4O2. Oxymethylene compounds, characterized by the grouping >C:CH(OH), also give benzene derivatives by hydrolytic condensation between three molecules; thus oxymethylene acetone, or formyl acetone, CH3·CO·CH:CH(OH), formed by acting on formic ester with acetone in the presence of sodium ethylate, readily yields [1.3.5]-triacetylbenzene, C6H3(CO·CH3)3; oxymethylene acetic ester or formyl acetic ester or β-oxyacrylic ester, (HO)CH:CH·CO2C2H5, formed by condensing acetic ester with formic ester, and also its dimolecular condensation product, coumalic acid, readily yields esters of [1.3.5]-benzene tricarboxylic acid or trimesic acid (see Ber., 1887, 20, p. 2930).
In 1890, O. Doebner (Ber. 23, p. 2377) investigated the condensation of pyroracemic acid, CH3·CO·COOH, with various aliphatic aldehydes, and obtained from two molecules of the acid and one of the aldehyde in the presence of baryta water alkylic isophthalic acids: with acetaldehyde [1.3.5]-methylisophthalic acid or uvitic acid, C6H3·CH3·(COOH)2, was obtained, with propionic aldehyde [1.3.5]-ethylisophthalic acid, and with butyric aldehyde the corresponding propylisophthalic acid. We may here mention the synthesis of oxyuvitic ester (5-methyl-4-oxy-1-3-benzene dicarboxylic ester) by the condensation of two molecules of sodium acetoacetic ester with one of chloroform (Ann., 1883, 222, p. 249). Of other syntheses of true benzene derivatives, mention may be made of the formation of orcinol or [3.5]-dioxytoluene from dehydracetic acid; and the formation of esters of oxytoluic acid (5-methyl-3-oxy-benzoic acid), C6H3·CH3·OH·COOH, when acetoneoxalic ester, CH3·CO·CH2·CO·CO·CO2C2H5, is boiled with baryta (Ber., 1889, 22, p. 3271). Of interest also are H.B. Hill and J. Torray’s observations on nitromalonic aldehyde, NO2·CH(CHO)2, formed by acting on mucobromic acid, probably CHO·CBr:CBr:COOH, with alkaline nitrites; this substance condenses with acetone to give p-nitrophenol, and forms [1.3.5]-trinitrobenzene when its sodium salt is decomposed with an acid.
By passing carbon monoxide over heated potassium J. von Liebig discovered, in 1834, an interesting aromatic compound, potassium carbon monoxide or potassium hexaoxybenzene, the nature of which was satisfactorily cleared up by R. Nietzki and T. Benckiser (Ber. 18, p. 499) in 1885, who showed that it yielded hexaoxybenzene, C6(OH)6, when acted upon with dilute hydrochloric acid; further investigation of this compound brought to light a considerable number of highly interesting derivatives (see Quinones). Another hexa-substituted benzene compound capable of direct synthesis is mellitic acid or benzene carboxylic acid, C6(COOH)6. This substance, first obtained from the mineral honeystone, aluminium mellitate, by M.H. Klaproth in 1799, is obtained when pure carbon (graphite or charcoal) is oxidized by alkaline permanganate, or when carbon forms the positive pole in an electrolytic cell (Ber., 1883, 16, p. 1209). The composition of this substance was determined by A. von Baeyer in 1870, who obtained benzene on distilling the calcium salt with lime.
Hitherto we have generally restricted ourselves to syntheses which result in the production of a true benzene ring; but there are many reactions by which reduced benzene rings are synthesized, and from the compounds so obtained true benzenoid compounds may be prepared. Of such syntheses we may notice: the condensation of sodium malonic ester to phloroglucin tricarboxylic ester, a substance which gives phloroglucin or trioxybenzene when fused with alkalis, and behaves both as a triketohexamethylene tricarboxylic ester and as a trioxybenzene tricarboxylic ester; the condensation of succinic ester, (CH2·CO2C2H5)2, under the influence of sodium to succinosuccinic ester, a diketohexamethylene dicarboxylic ester, which readily yields dioxyterephthalic acid and hydroquinpne (F. Herrmann, Ann., 1882, 211, p. 306; also see below, Configuration of the Benzene Complex); the condensation of acetone dicarboxylic ester with malonic ester to form triketohexamethylene dicarboxylic ester (E. Rimini, Gazz. Chem., 1896, 26, (2), p. 374); the condensation of acetone-di-propionic acid under the influence of boiling water to a diketohexamethylene propionic acid (von Pechmann and Sidgwick, Ber., 1904, 37, p. 3816). Many diketo compounds suffer condensation between two molecules to form hydrobenzene derivatives, thus α, γ-di-acetoglutaric ester, C2H5O2C(CH3·CO)CH·CH2·CH(CO·CH3)CO2C2H5, yields a methyl-ketohexamethylene, while γ-acetobutyric ester, CH3CO(CH2)2CO2C2H5, is converted into dihydroresorcinol or m-diketohexamethylene by sodium ethylate; this last reaction is reversed by baryta (see Decompositions of Benzene Ring). For other syntheses of hexamethylene derivatives, see Polymethylenes.
Decompositions of the Benzene Ring.—We have previously alluded to the relative stability of the benzene complex; consequently reactions which lead to its disruption are all the more interesting, and have engaged the attention of many chemists. If we accept Kekulé’s formula for the benzene nucleus, then we may expect the double linkages to be opened up partially, either by oxidation or reduction, with the formation of di-, tetra-, or hexa-hydro derivatives, or entirely, with the production of open chain compounds. Generally rupture occurs at more than one point; and rarely are the six carbon atoms of the complex regained as an open chain. Certain compounds withstand ring decomposition much more strongly than others; for instance, benzene and its homologues, carboxylic acids, and nitro compounds are much more stable towards oxidizing agents than amino- and oxy-benzenes, aminophenols, quinones, and oxy-carboxylic acids.
Strong oxidation breaks the benzene complex into such compounds as carbon dioxide, oxalic acid, formic acid, &c.; such decompositions are of little interest. More important are Kekulé’s Simple oxidation. observations that nitrous acid oxidizes pyrocatechol or [1.2]-dioxybenzene, and protocatechuic acid or [3.4]-dioxybenzoic acid to dioxytartaric acid, (C(OH)2·COOH)2 (Ann., 1883, 221, p. 230); and O. Doebner’s preparation of mesotartaric acid, the internally compensated tartaric acid, (CH(OH)·COOH)2, by oxidizing phenol with dilute potassium permanganate (Ber., 1891, 24, p. 1753).
For many years it had been known that a mixture of potassium chlorate and hydrochloric or sulphuric acids possessed strong oxidizing powers. L. Carius showed that potassium Chlorination and oxidation. chlorate and sulphuric acid oxidized benzene to trichlor-phenomalic acid, a substance afterwards investigated by Kekulé and O. Strecker (Ann., 1884, 223, p. 170), and shown to be β-trichloracetoacrylic acid, CCl3·CO·CH:CH·COOH, which with baryta gave chloroform and maleic acid. Potassium chlorate and hydrochloric acid oxidize phenol, salicylic acid (o-oxybenzoic acid), and gallic acid ([2.3.4] trioxybenzoic acid) to trichlorpyroracemic acid (isotrichlorglyceric acid), CCl3·C(OH)2·CO2H, a substance also obtained from trichloracetonitrile, CCl3·CO·CN, by hydrolysis. We may also notice the conversion of picric acid, ([2.4.6]-trinitrophenol) into chloropicrin, CCl3NO2, by bleaching lime (calcium hypochlorite), and into bromopicrin, CBr3NO2, by bromine water.
The action of chlorine upon di-and tri-oxybenzenes has been carefully investigated by Th. Zincke; and his researches have led to the discovery of many chlorinated oxidation products which admit of decomposition into cyclic compounds containing fewer carbon atoms than characterize the benzene ring, and in turn yielding open-chain or aliphatic compounds. In general, the rupture occurs between a-keto group (CO) and a keto-chloride group (CCl2), into which two adjacent carbon atoms of the ring are converted by the oxidizing and substituting action of chlorine. Decompositions of this nature were first discovered in the naphthalene series, where it was found that derivatives of indene (and of hydrindene and indone) and also of benzene resulted; Zincke then extended his methods to the disintegration of the oxybenzenes and obtained analogous results, R-pentene and aliphatic derivatives being formed (R- symbolizing a ringed nucleus).When treated with chlorine, pyrocatechol (1.2 or ortho-dioxybenzene) (1) yields a tetrachlor ortho-quinone, which suffers further chlorination to hexachlor-o-diketo-R-hexene (2). This substance is transformed into hexachlor-R-pentene oxycarboxylic acid (3) when digested with water; and chromic acid oxidizes this substance to hexachlor-R-pentene (4). The ring of this compound is ruptured by caustic soda with the formation of perchlorvinyl acrylic acid (5), which gives on reduction ethidine propionic acid (6), a compound containing five of the carbon atoms originally in the benzene ring (see Zincke, Ber., 1894, 27, p. 3364) (the carbon atoms are omitted in some of the formulae).
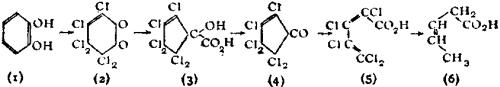
Resorcin (1.3 or meta dioxybenzene) (1) is decomposed in a somewhat similar manner. Chlorination in glacial acetic acid solution yields pentachlor-m-diketo-R-hexene (2) and, at a later stage, heptachlor-m-diketo-R-hexene (3). These compounds are both decomposed by water, the former giving dichloraceto-trichlor-crotonic acid (4), which on boiling with water gives dichlormethyl-vinyl-a-diketone (5). The heptachlor compound when treated with chlorine water gives trichloraceto-pentachlorbutyric acid (6), which is hydrolysed by alkalis to chloroform and pentachlorglutaric acid (7), and is converted by boiling water into tetrachlor-diketo-R-pentene (8). This latter compound may be chlorinated to perchloracetoacrylic chloride (9), from which the corresponding acid (10) is obtained by treatment with water; alkalis hydrolyse the acid to chloroform and dichlormaleic acid (11).
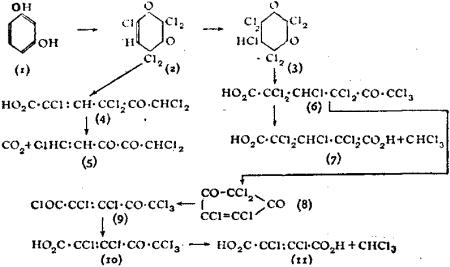
Hydroquinone (1.4 or para-dioxybenzene) (1) gives with chlorine, first, a tetrachlorquinone (2), and then hexachlor-p-diketo-R-hexene (3), which alcoholic potash converts into perchloracroylacrylic acid (4). This substance, and also the preceding compound, is converted by aqueous caustic soda into dichlormaleic acid, trichlorethylene, and hydrochloric acid (5) (Th. Zincke and O. Fuchs, Ann., 1892, 267, p. 1).
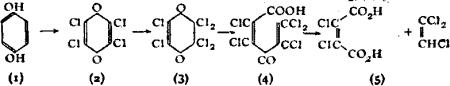
Phloroglucin (1.3.5-trioxybenzene) (1) behaves similarly to resorcin, hexachlor [1.3.5] triketo-R-hexylene (2) being first formed. This compound is converted by chlorine water into octachloracetylacetone (3); by methyl alcohol into the ester of dichlormalonic acid and tetrachloracetone (4); whilst ammonia gives dichloracetamide (5) (Th. Zincke and O. Kegel, Ber., 1890, 23, p. 1706).

When phenol is oxidized in acid solution by chlorine, tetrachlorquinone is obtained, a compound also obtainable from hydroquinone. By conducting the chlorination in alkaline solution, Reduction in alkaline solution. A. Hantzsch (Ber., 1889, 22, p. 1238) succeeded in obtaining derivatives of o-diketo-R-hexene, which yield R-pentene and aliphatic compounds on decomposition. When thus chlorinated phenol (1) yields trichlor-o-diketo-R-hexene (2), which may be hydrolysed to an acid (3), which, in turn, suffers rearrangement to trichlor-R-pentene-oxycarboxylic acid (4). Bromine water oxidizes this substance to oxalic acid and tetrabrom-dichloracetone (5).
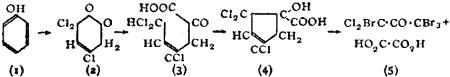
The reduction of o-oxybenzoic acids by sodium in amyl alcohol solution has been studied by A. Einhorn and J.S. Lumsden (Ann., 1895, 286, p. 257). It is probable that tetrahydro acids are first formed, which suffer rearrangement to orthoketone carboxylic acids. These substances absorb water and become pimelic acids. Thus salicylic acid yields n-pimelic acid, HOOC·(CH2)5·COOH, while o-, m-, and p-cresotinic acids, C6H3(CH3)(OH)(COOH), yield isomeric methylpimelic acids.
Resorcin on reduction gives dihydroresorcin, which G. Merling (Ann., 1894, 278, p. 20) showed to be converted into n-glutaric acid, HOOC·(CH2)3·COOH, when oxidized with potassium permanganate; according to D. Vörlander (Ber., 1895, 28, p. 2348) it is converted into γ-acetobutyric acid, CH3CO·(CH2)3·COOH, when heated with baryta to 150-160°.
Configuration of the Benzene Complex.—The development of the “structure theory” in about 1860 brought in its train an appreciation of the chemical structure of the derivatives of benzene. The pioneer in this field was August Kekulé, who, in 1865 (Ann., 137, p. 129; see also his Lehrbuch der organischen Chemie), submitted his well-known formula for benzene, so founding the “benzene theory” and opening up a problem which, notwithstanding the immense amount of labour since bestowed upon it, still remains imperfectly solved. Arguing from the existence of only one mono-substitution derivative, and of three di-derivatives (statements of which the rigorous proof was then wanting), he was led to arrange the six carbon atoms in a ring, attaching a hydrogen atom to each carbon atom; being left with the fourth carbon valencies, he mutually saturated these in pairs, thus obtaining the symbol I (see below). The value of this ringed structure was readily perceived, but objections were raised with respect to Kekulé’s disposal of the fourth valencies. In 1866 Sir James Dewar proposed an unsymmetrical form (II); while in 1867, A. Claus (Theoretische Betrachtungen und deren Anwendung zur Systematik der organischen Chemie) proposed his diagonal formula (III), and two years later, A. Ladenburg (Ber., 2, p. 140) devised his prism formula (IV), the six carbon atoms being placed at the six corners of a right equilateral triangular prism, with its plane projections (V, VI).
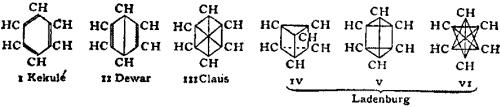
One of the earliest and strongest objections urged against Kekulé’s formula was that it demanded two isomeric ortho-di-substitution derivatives; for if we number the carbon atoms in cyclical Objections to Kekulé’s formula. order from 1 to 6, then the derivatives 1·2 and 1·6 should be different.13 Ladenburg submitted that if the 1·2 and 1·6 compounds were identical, then we should expect the two well-known crotonic acids, CH3·CH:CH·COOH and CH2:CH·CH2·COOH, to be identical. This view was opposed by Victor Meyer and Kekulé. The former pointed out that the supposed isomerism was not due to an arrangement of atoms, but to the disposition of a valency, and therefore it was doubtful whether such a subtle condition could exert any influence on the properties of the substance. Kekulé answered Ladenburg by formulating a dynamic interpretation of valency. He assumed that if we have one atom connected by single bonds to (say) four other atoms, then in a certain unit of time it will collide with each of these atoms in turn. Now suppose two of the attached atoms are replaced by one atom, then this atom must have two valencies directed to the central atom; and consequently, in the same unit of time, the central atom will collide once with each of the two monovalent atoms and twice with the divalent. Applying this notion to benzene, let us consider the impacts made by the carbon atom (1) which we will assume to be doubly linked to the carbon atom (2) and singly linked to (6), h standing for the hydrogen atom. In the first unit of time, the impacts are 2, 6, h, 2; and in the second 6, 2, h, 6. If we represent graphically the impacts in the second unit of time, we perceive that they point to a configuration in which the double linkage is between the carbon atoms 1 and 6, and the single linkage between 1 and 2. Therefore, according to Kekulé, the double linkages are in a state of continual oscillation, and if his dynamical notion of valency, or a similar hypothesis, be correct, then the difference between the 1.2 and 1.6 di-derivatives rests on the insufficiency of his formula, which represents the configuration during one set of oscillations only. The difference is only apparent, not real. An analogous oscillation prevails in the pyrazol nucleus, for L. Knorr (Ann., 1894, 279, p. 188) has shown that 3- and 5-methylpyrazols are identical.
The explanation thus attempted by Kekulé was adversely criticized, more especially by A. Ladenburg, who devoted much attention to the study of the substitution products of benzene, and Ladenburg’s formula. to the support of his own formula. His views are presented in his Pamphlet: Theorie der aromatischen Verbindungen, 1876. The prism formula also received support from the following data: protocatechuic acid when oxidized by nitrous acid gives carboxytartronic acid, which, on account of its ready decomposition into carbon dioxide and tartronic acid, was considered to be HO·C(COOH)3. This implied that in the benzene complex there was at least one carbon atom linked to three others, thus rendering Kekulé’s formula impossible and Ladenburg’s and Claus’ possible. Kekulé (Ann., 1883, 221, p. 230), however, reinvestigated this acid; he showed that it was dibasic and not tribasic; that it gave tartaric acid on reduction; and, finally, that it was dioxytartaric acid, HOOC·C(OH)2·C(OH)2·COOH. The formation of this substance readily follows from Kekulé’s formula, while considerable difficulties are met with when one attempts an explanation based on Ladenburg’s representation. Kekulé also urged that the formation of trichlorphenomalic acid, shown by him and O. Strecker to be trichloracetoacrylic acid, was more favourably explained by his formula than by Ladenburg’s.
Other objections to Ladenburg’s formula resulted from A. von Baeyer’s researches (commenced in 1886) on the reduced phthalic acids. Baeyer pointed out that although benzene derivatives Baeyer’s researches. were obtainable from hexamethylene compounds, yet it by no means follows that only hexamethylene compounds need result when benzene compounds are reduced. He admitted the possibility of the formulae of Kekulé, Claus, Dewar and Ladenburg, although as to the last di-trimethylene derivatives should be possible reduction products, being formed by severing two of the prism edges; and he attempted to solve the problem by a systematic investigation of the reduced phthalic acids.
Ladenburg’s prism admits of one mono-substitution derivative and three di-derivatives. Furthermore, it is in accordance with certain simple syntheses of benzene derivatives (e.g. from acetylene and acetone); but according to Baeyer (Ber., 1886, 19, p. 1797) it fails to explain the formation of dioxyterephthalic ester from succinosuccinic ester, unless we make the assumption that the transformation of these substances is attended by a migration of the substituent groups. For succinosuccinic ester, formed by the action of sodium on two molecules of succinic ester, has either of the formulae (I) or (II); oxidation of the free acid gives dioxyterephthalic acid in which the para-positions must remain substituted as in (I) and (II). By projecting Ladenburg’s prism on a plane and numbering the atoms so as to correspond with Kekulé’s form, viz. that 1.2 and 1.6 should be ortho-positions, 1.3 and 1.5 meta-, and 1.4 para-, and following out the transformation on the Ladenburg formula, then an ortho-dioxyterephthalic acid (IV) should result, a fact denied by experience, and inexplicable unless we assume a wandering of atoms. Kekulé’s formula (III), on the other hand, is in full agreement (Baeyer). This explanation has been challenged by Ladenburg
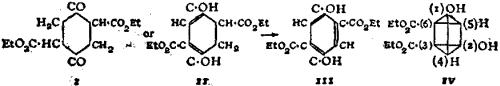
(Ber., 1886, 19, p. 971; Ber., 1887, 20, p. 62) and by A.K. Miller (J.C.S. Trans., 1887, p. 208). The transformation is not one of the oxidation of a hexamethylene compound to a benzenoid compound, for only two hydrogen atoms are removed. Succinosuccinic ester behaves both as a ketone and as a phenol, thereby exhibiting desmotropy; assuming the ketone formula as indicating the constitution, then in Baeyer’s equation we have a migration of a hydrogen atom, whereas to bring Ladenburg’s formula into line, an oxygen atom must migrate.
The relative merits of the formulae of Kekulé, Claus and Dewar were next investigated by means of the reduction products of benzene, it being Baeyer’s intention to detect whether double linkages were or were not present in the benzene complex.
To follow Baeyer’s results we must explain his nomenclature of the reduced benzene derivatives. He numbers the carbon atoms placed at the corners of a hexagon from 1 to 6, and each side in the same order, so that the carbon atoms 1 and 2 are connected by the side 1, atoms 2 and 3 by the side 2, and so on. A doubly linked pair of atoms is denoted by the sign Δ with the index corresponding to the side; if there are two pairs of double links, then indices corresponding to both sides are employed. Thus Δ1 denotes a tetrahydro derivative in which the double link occupies the side 1; Δ1.3, a dihydro derivative, the double links being along the sides 1 and 3. Another form of isomerism is occasioned by spatial arrangements, many of the reduced terephthalic acids existing in two stereo-isomeric forms. Baeyer explains this by analogy with fumaric and maleic acids: he assumes the reduced benzene ring to lie in a plane; when both carboxyl groups are on the same side of this plane, the acids, in general, resemble maleic acids, these forms he denotes by Γcis-cis, or shortly cis-; when the carboxyl groups are on opposite sides, the acids correspond to fumaric acid, these forms are denoted by Γcis-trans, or shortly trans-.
By reducing terephthalic acid with sodium amalgam, care being taken to neutralize the caustic soda simultaneously formed by passing in carbon dioxide, Δ2.5 dihydroterephthalic acid is obtained; this results from the splitting of a para-linkage. By boiling with water the Δ2.5 acid is converted into the Δ1.5 dihydroterephthalic acid. This acid is converted into the Δ1.4 acid by soda, and into the Δ2 tetrahydro acid by reduction. From this acid the Δ1.3 dihydro and the Δ1 tetrahydro acids may be obtained, from both of which the hexahydro acid may be prepared. From these results Baeyer concluded that Claus’ formula with three para-linkings cannot possibly be correct, for the Δ2.5 dihydroterephthalic acid undoubtedly has two ethylene linkages, since it readily takes up two or four atoms of bromine, and is oxidized in warm aqueous solution by alkaline potassium permanganate. But the formation of the Δ2.5 acid as the first reduction product is not fully consistent with Kekulé’s symbol, for we should then expect the Δ1.3 or the Δ1.5 acid to be first formed (see also Polymethylenes).
The stronger argument against the ethylenoid linkages demanded by Kekulé’s formula is provided by the remarkable stability towards oxidizing and reducing agents which characterizes all benzenoid compounds. From the fact that reduction products containing either one or two double linkages behave exactly as unsaturated aliphatic compounds, being readily reduced or oxidized, and combining with the halogen elements and haloid acids, it seems probable that in benzenoid compounds the fourth valencies are symmetrically distributed in such a manner as to induce a peculiar stability in the molecule. Such a configuration was proposed in 1887 by H.E. Armstrong (J.C.S. Trans., 1887, p. 258), and shortly afterwards by Baeyer (Ann., 1888, 245, p. 103). In this formula, the so-called “centric formula,” the assumption made is that the fourth valencies are simply directed towards the centre of the ring; nothing further is said about the fourth valencies except that they exert a pressure towards the centre. Claus maintained that Baeyer’s view was identical with his own, for as in Baeyer’s formula, the fourth valencies have a different function from the peripheral valencies, being united at the centre in a form of potential union.
It is difficult to determine which configuration most accurately explains the observed facts; Kekulé’s formula undoubtedly explains the synthetical production of benzenoid compounds most satisfactorily, and W. Marckwald (Ann., 1893, 274, p. 331; 1894, 279, p. 14) has supported this formula from considerations based on the syntheses of the quinoline ring. Further researches by Baeyer, and upon various nitrogenous ring systems by E. Bamberger (a strong supporter of the centric formula), have shown that the nature of the substituent groups influences the distribution of the fourth valencies; therefore it may be concluded that in compounds the benzene nucleus appears to be capable of existence in two tautomeric forms, in the sense that each particular derivative possesses a definite constitution. The benzene nucleus presents a remarkable case, which must be considered in the formulation of any complete theory of valency. From a study of the reduction of compounds containing two ethylenic bonds united by a single bond, termed a “conjugated system,” E. Thiele suggested a doctrine of “partial valencies,” which assumes that in addition to the ordinary valencies, each doubly linked atom has a partial valency, by which the atom first interacts. When applied to benzene, a twofold conjugated system is suggested in which the partial valencies of adjacent atoms neutralize, with the formation of a potential double link. The stability of benzene is ascribed to this conjugation.14
Physico-chemical properties have also been drawn upon to decide whether double unions are present in the benzene complex; but here the predilections of the observers Physico-chemical methods. apparently influence the nature of the conclusions to be drawn from such data. It is well known that singly, doubly and trebly linked carbon atoms affect the physical properties of substances, such as the refractive index, specific volume, and the heat of combustion; and by determining these constants for many substances, fairly definite values can be assigned to these groupings. The general question of the relation of the refractive index to constitution has been especially studied by J.W. Brühl, who concluded that benzene contained 3 double linkages; whereas, in 1901, Pellini (Gazetta, 31, i. p. 1) calculated that 9 single linkages were present. A similar contradiction apparently exists with regard to the specific volume, for while benzene has a specific volume corresponding to Claus’ formula, toluene, or methylbenzene, rather points to Kekulé’s. The heat of combustion, as first determined by Julius Thomsen, agreed rather better with the presence of nine single unions. His work was repeated on a finer scale by M.P.E. Berthelot of Paris, and F.C.A. Stohmann of Leipzig; and the new data and the conclusions to be drawn from them formed the subject of much discussion, Brühl endeavouring to show how they supported Kekulé’s formula, while Thomsen maintained that they demanded the benzene union to have a different heat of combustion from the acetylene union. Thomsen then investigated heats of combustion of various benzenoid hydrocarbons—benzene, naphthalene, anthracene, phenanthrene, &c.—in the crystallized state. It was found that the results were capable of expression by the empirical relation CaH2b = 104.3b + 49.09m + 105.47n, where CaH2b denotes the formula of the hydrocarbon, m the number of single carbon linkings and n the number of double linkings, m and n being calculated on the Kekulé formulae. But, at the same time, the constants in the above relation are not identical with those in the corresponding relation empirically deduced from observations on fatty hydrocarbons; and we are therefore led to conclude that a benzene union is considerably more stable than an ethylene union.
Mention may be made of the absorption spectrum of benzene. According to W.N. Hartley (J.C.S., 1905, 87, p. 1822), there are six bands in the ultra-violet, while E.C.C. Baly and J.N. Collie (J.C.S., 1905, 87, p. 1332; 1906, 89, p. 524) record seven. These bands are due to molecular oscillations; Hartley suggests the carbon atoms to be rotating and forming alternately single and double linkages, the formation of three double links giving three bands, and of three single links another three; Baly and Collie, on the other hand, suggest the making and breaking of links between adjacent atoms, pointing out that there are seven combinations of one, two and three pairs of carbon atoms in the benzene molecule.
Stereo-chemical Configurations.—Simultaneously with the discussions of Kekulé, Ladenburg, Claus, Baeyer and others as to the merits of various plane formulae of the benzene complex, there were published many suggestions with regard to the arrangement of the atoms in space, all of which attempted to explain the number of isomers and the equivalence of the hydrogen atoms. The development of stereo-isomerism at the hands of J. Wislicenus, Le Bel and van ’t Hoff has resulted in the introduction of another condition which formulae for the benzene complex must satisfy, viz. that the hydrogen atoms must all lie in one plane. The proof of this statement rests on the fact that if the hydrogen atoms were not co-planar, then substitution derivatives (the substituting groups not containing asymmetric carbon atoms) should exist in enantiomorphic forms, differing in crystal form and in their action on polarized light; such optical antipodes have, however, not yet been separated. Ladenburg’s prism formula would give two enantiomorphic ortho-di-substitution derivatives; while forms in which the hydrogen atoms are placed at the corners of a regular octahedron would yield enantiomorphic tri-substitution derivatives.
The octahedral formula discussed by Julius Thomsen (Ber., 1886, 19, p. 2944) consists of the six carbon atoms placed at the corners of a regular octahedron, and connected together by the full lines as shown in (I); a plane projection gives a hexagon with diagonals (II). Reduction to hexamethylene compounds necessitates the disruption of three of the edges of the octahedron, the diagonal linkings remaining intact, or, in the plane projection, three peripheral linkages, the hexamethylene ring assuming the form (III):
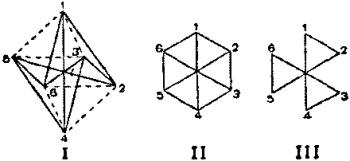
In 1888 J.E. Marsh published a paper (Phil. Mag. [V.], 26, p. 426) in which he discussed various stereo-chemical representations of the benzene nucleus. (The stereo-chemistry of carbon compounds has led to the spatial representation of a carbon atom as being situated at the centre of a tetrahedron, the four valencies being directed towards the apices; see above, and Isomerism.) A form based on Kekulé’s formula consists in taking three pairs of tetrahedra, each pair having a side in common, and joining them up along the sides of a regular hexagon by means of their apices. This form, afterwards supported by Carl Graebe (Ber., 1902, 35, p. 526; see also Marsh’s reply, Journ. Chem. Soc. Trans., 1902, p. 961) shows the proximity of the ortho-positions, but fails to explain the identity of 1·2 and 1·6 compounds. Arrangements connected with Claus’ formula are obtained by placing six tetrahedra on the six triangles formed by the diagonals of a plane hexagon. The form in which the tetrahedra are all on one side, afterwards discussed by J. Loschmidt (Monats., 1890, II, p. 28), would not give stereo-isomers; and the arrangement of placing the tetrahedra on alternate sides, a form afterwards developed by W. Vaubel (Journ. Pr. Chem., 1894[2], 49, p. 308), has the advantage of bringing the meta-positions on one side, and the ortho- and para- on opposite sides, thus exhibiting the similarity actually observed between these series of compounds. Marsh also devised a form closely resembling that of Thomsen, inasmuch as the carbon atoms occupied the angles of a regular octahedron, and the diagonal linkages differed in nature from the peripheral, but differing from Thomsen’s since rupture of the diagonal and not peripheral bonds accompanied the reduction to hexamethylene.
We may also notice the model devised by H. Sachse (Ber., 1888, 21, 2530; Zeit. fur phys. Chem., II, p. 214; 23, p. 2062). Two parallel triangular faces are removed from a cardboard model of a regular octahedron, and on the remaining six faces tetrahedra are then placed; the hydrogen atoms are at the free angles. This configuration is, according to Sachse, more stable than any other form; no oscillation is possible, the molecule being only able to move as a whole. In 1897, J.N. Collie (Journ. Chem. Soc. Trans., p. 1013) considered in detail an octahedral form, and showed how by means of certain simple rotations of his system the formulae of Kekulé and Claus could be obtained as projections. An entirely new device, suggested by B. König (Chem. Zeit., 1905, 29, p. 30), assumed the six carbon atoms to occupy six of the corners of a cube, each carbon atom being linked to a hydrogen atom and by single bonds to two neighbouring carbon atoms, the remaining valencies being directed to the unoccupied corners of the cube, three to each, where they are supposed to satisfy each other.
Condensed Nuclei.
Restricting ourselves to compounds resulting from the fusion of benzene rings, we have first to consider naphthalene, C10H8, which consists of two benzene rings having a pair of carbon atoms in common. The next members are the isomers anthracene and phenanthrene, C14H10, formed from three benzene nuclei. Here we shall only discuss the structure of these compounds in the light of the modern benzene theories; reference should be made to the articles Naphthalene, Anthracene and Phenanthrene for syntheses, decompositions, &c.
Naphthalene.—Of the earlier suggestions for the constitution of naphthalene we notice the formulae of Wreden (1) and (2), Berthelot and Balls (3), R.A.C.E. Erlenmeyer (4) and Adolf Claus (5).
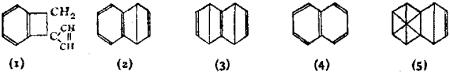
The first suggestion is quite out of the question. C. Graebe in 1866 (Ann. 149, p. 20) established the symmetry of the naphthalene nucleus, and showed that whichever half of the molecule be oxidized the same phthalic acid results. Therefore formula (2), being unsymmetrical, is impossible. The third formula is based on Dewar’s benzene formula, which we have seen to be incorrect. Formula (4) is symmetrical and based on Kekulé’s formula: it is in full accord with the syntheses and decompositions of the naphthalene nucleus and the number of isomers found. In 1882 Claus suggested a combination of his own and Dewar’s benzene formulae. This is obviously unsymmetrical, consisting of an aliphatic and an aromatic nucleus; Claus explained the formation of the same phthalic acid from the oxidation of either nucleus by supposing that if the aromatic group be oxidized, the aliphatic residue assumes the character of a benzene nucleus. Bamberger opposed Claus’ formula on the following grounds:—The molecule of naphthalene is symmetrical, since 2.7 dioxynaphthalene is readily esterified by methyl iodide and sulphuric acid to a dimethyl ether; and no more than two mono-substitution derivatives are known. The molecule is aromatic but not benzenoid; however, by the reduction of one half of the molecule, the other assumes a benzenoid character.
If β-naphthylamine and β-naphthol be reduced, tetrahydro products are obtained in which the amino- or oxy-bearing half of the molecule becomes aliphatic in character. The compounds so obtained, alicyclic-β-tetrahydronaphthylamine and alicyclic-β-tetrahydronaphthol, closely resemble β-aminodiethylbenzene, C6H4(C2H5)·C2H4NH2, and β-oxydiethylbenzene, C6H4(C2H5)·C2H4OH. If α-naphthylamine and α-naphthol be reduced, the hydrogen atoms attach themselves to the non-substituted half of the molecule, and the compounds so obtained resemble aminodiethylbenzene, C6H3·NH2(C2H5)2 and oxydiethylbenzene, C6H3·OH(C2H5)2. Bamberger’s observations on reduced quinoline derivatives point to the same conclusion, that condensed nuclei are not benzenoid, but possess an individual character, which breaks down, however, when the molecule is reduced.
It remains, therefore, to consider Erlenmeyer’s formula and those derived from the centric hypothesis. The former, based on Kekulé’s symbol for benzene, explains the decompositions and syntheses of the ring, but the character of naphthalene is not in keeping with the presence of five double linkages, although it is more readily acted upon than benzene is. On the centric hypothesis two formulae are possible: (i) due to H.E. Armstrong, and (2) due to E. Bamberger.
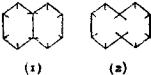
In the first symbol it is assumed that one of the affinities of each of the two central carbon atoms common to the two rings acts into both rings, an assumption involving a somewhat wide departure from all ordinary views as to the manner in which affinity acts. This symbol harmonizes with the fact that the two rings are in complete sympathy, the one responding to every change made in the other. Then, on account of the relatively slight—because divided—influence which would be exercised upon the two rings by the two affinities common to both, the remaining four centric affinities of each ring would presumably be less attracted into the ring than in the case of benzene; consequently they would be more active outwards, and combination would set in more readily. When, as in the formation of naphthalene tetrachloride, for example, the one ring becomes saturated, the other might be expected to assume the normal centric form and become relatively inactive. This is absolutely the case. On the other hand, if substitution be effected in the one ring, and the affinities in that ring become attracted inwards, as apparently happens in the case of benzene, the adjoining ring should become relatively more active because the common affinities would act less into it. Hence, unless the radical introduced be one which exercises a special attractive influence, substitution should take place in preference in the previously unsubstituted ring. In practice this usually occurs; for example, on further bromination, α-bromonaphthalene yields a mixture of the (1.4) and (1.5) dibromonaphthalenes; and when nitronaphthalene is either brominated, or nitrated or sulphonated, the action is practically confined to the second ring. The centric formula proposed by Bamberger represents naphthalene as formed by the fusion of two benzene rings, this indicates that it is a monocyclic composed of ten atoms of carbon. The formula has the advantage that it may be constructed from tetrahedral models of the carbon atom; but it involves the assumption that the molecule has within it a mechanism, equivalent in a measure to a system of railway points, which can readily close up and pass into that characteristic of benzene.
Anthracene and Phenanthrene.—These isomeric hydrocarbons, of the formula C14H10, are to be regarded as formed by the fusion of three benzenoid rings as represented by the symbols:—
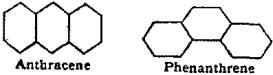
In both cases the medial ring is most readily attacked; and various formulae have been devised which are claimed by their authors to represent this and other facts. According to Armstrong, anthracene behaves unsymmetrically towards substituents, and hence one lateral ring differs from the other; he represents the molecule as consisting of one centric ring, the remaining medial and lateral ring being ethenoid. Bamberger, on the other hand, extends his views on benzene and naphthalene and assumes the molecule to be (1). For general purposes, however, the symbol (2), in which the lateral rings are benzenoid and the medial ring fatty, represents quite adequately the syntheses, decompositions, and behaviour of anthracene.
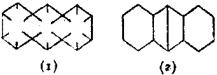
Phenanthrene is regarded by Armstrong as represented by (3), the lateral rings being benzenoid, and the medial ring fatty; Bamberger, however, regards it as (4), the molecule being entirely aromatic. An interesting observation by Baeyer, viz. that stilbene, C6H5·CH:CH·C6H5, is very readily oxidized, while phenanthrene is not, supports, in some measure, the views of Bamberger.
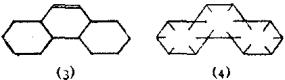
Heterocyclic Compounds.
During recent years an immense number of ringed or cyclic compounds have been discovered, which exhibit individual characters more closely resembling benzene, naphthalene, &c. than purely aliphatic substances, inasmuch as in general they contain double linkages, yet withstand oxidation, and behave as nuclei, forming derivatives in much the same way as benzene. By reduction, the double linkages become saturated, and compounds result which stand in much about the same relation to the original nucleus as hexamethylene does to benzene. In general, therefore, it may be considered that the double linkages are not of exactly the same nature as the double linkage present in ethylene and ethylenoid compounds, but that they are analogous to the potential valencies of benzene. The centric hypothesis has been applied to these rings by Bamberger and others; but as in the previous rings considered, the ordinary representation with double and single linkages generally represents the syntheses, decompositions, &c.; exceptions, however, are known where it is necessary to assume an oscillation of the double linkage. Five- and six-membered rings are the most stable and important, the last-named group resulting from the polymerization of many substances; three- and four-membered rings are formed with difficulty, and are easily ruptured; rings containing seven or more members are generally unstable, and are relatively little known. The elements which go to form heterocyclic rings, in addition to carbon, are oxygen, sulphur, selenium and nitrogen. It is remarkable that sulphur can replace two methine or CH groups with the production of compounds greatly resembling, the original one. Thus benzene, (CH)6, gives thiophene, (CH)4S, from which it is difficultly distinguished; pyridine, (CH)5N, gives thiazole, (CH)3·N·S, which is a very similar substance; naphthalene gives thionaphthen, C8H6S, with which it shows great analogies, especially in the derivatives. Similarly a CH group may be replaced by a nitrogen atom with the production of compounds of similar stability; thus benzene gives pyridine, naphthalene gives quinoline and isoquinoline; anthracene gives acridine and α and β anthrapyridines. Similarly, two or more methine groups may be replaced by the same number of nitrogen atoms with the formation of rings of considerable stability.
Most of the simple ring systems which contain two adjacent carbon atoms may suffer fusion with any other ring (also containing two adjacent carbon atoms) with the production of nuclei of greater complexity. Such condensed nuclei are, in many cases, more readily obtained than the parent nucleus. The more important types are derived from aromatic nuclei, benzene, naphthalene, &c.; the ortho-di-derivatives of the first named, lending themselves particularly to the formation of condensed nuclei. Thus ortho-phenylene diamine yields the following products:—
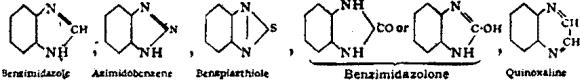
In some cases oxidation of condensed benzenoid-heterocyclic nuclei results in the rupture of the heterocyclic ring with the formation of a benzene dicarboxylic acid; but if the aromatic nucleus be weakened by the introduction of an amino group, then it is the benzenoid nucleus which is destroyed and a dicarboxylic acid of the heterocyclic ring system obtained.
Heterocyclic rings may be systematically surveyed from two aspects: (1) by arranging the rings with similar hetero-atoms according to the increasing number of carbon atoms, the so-called “homologous series”; or (2) by first dividing the ring systems according to the number of members constituting the ring, and then classifying these groups according to the nature of the hetero-atoms, the so-called “isologous series.” The second method possesses greater advantages, for rings of approximate stability come in one group, and, consequently, their derivatives may be expected to exhibit considerable analogies.
As a useful preliminary it is convenient to divide heterocyclic ring systems into two leading groups: (1) systems resulting from simple internal dehydration (or similar condensations) of saturated aliphatic compounds—such compounds are: the internal anhydrides or cyclic ethers of the glycols and thioglycols (ethylene oxide, &c.); the cyclic alkyleneimides resulting from the splitting off of ammonia between the amino groups of diamino-paraffins (pyrrolidine, piperazine, &c.); the cyclic esters of oxycarboxylic acids (lactones, lactides); the internal anhydrides of aminocarboxylic acids (lactams, betaines); cyclic derivatives of dicarboxylic acids (anhydrides, imides, alkylen-esters, alkylen-amides, &c.). These compounds retain their aliphatic nature, and are best classified with open-chain compounds, into which, in general, they are readily converted. (2) Systems which are generally unsaturated compounds, often of considerable stability, and behave as nuclei; these compounds constitute a well-individualized class exhibiting closer affinities to benzenoid substances than to the open-chain series.
The transition between the two classes as differentiated above may be illustrated by the following cyclic compounds, each of which contains a ring composed of four carbon atoms and one oxygen atom:
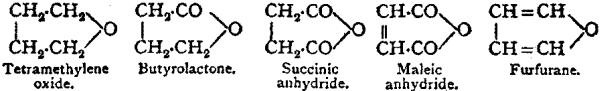
The first four substances are readily formed from, and converted into, the corresponding dihydroxy open-chain compound; these substances are truly aliphatic in character. The fifth compound, on the other hand, does not behave as an unsaturated aliphatic compound, but its deportment is that of a nucleus, many substitution derivatives being capable of synthesis. Reduction, however, converts it into an aliphatic compound. This is comparable with the reduction of the benzene nucleus into hexamethylene, a substance of an aliphatic character.
True ring systems, which possess the characters of organic nuclei, do not come into existence in three-and four-membered rings, their first appearance being in penta-atomic rings. The three primary members are furfurane, thiophene and pyrrol, each of which contains four methine or CH groups, and an oxygen, sulphur and imido (NH) member respectively; a series of compounds containing selenium is also known. The formulae of these substances are:
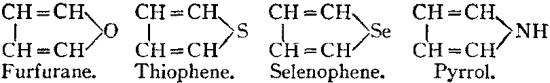
By substituting one or more CH groups in these compounds by nitrogen atoms, ring-systems, collectively known as azoles, result. Obviously, isomeric ring-systems are possible, since the carbon atoms in the original rings are not all of equal value. Thus furfurane yields the following rings by the introduction of one and two nitrogen atoms:
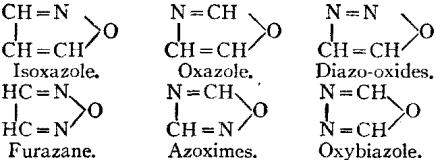
Thiophene yields a similar series: isothiazole (only known as the condensed ring, isobenzothiazole), thiazole, diazosulphides, piazthioles, azosulphimes and thiobiazole (the formulae are easily derived from the preceding series by replacing oxygen by sulphur). Thiophene also gives rise to triazsulphole, three nitrogen atoms being introduced. Selenophene gives the series: selenazole, diazoselenide and piaselenole, corresponding to oxazole, diazo-oxides and furazane. Pyrrol yields an analogous series: pyrazole, imidazole or glyoxaline, azimide or osotriazole, triazole and tetrazole:
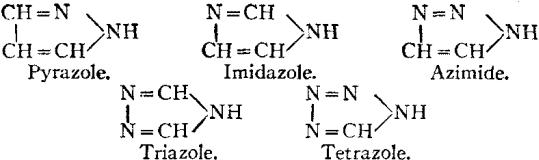
Six-membered ring systems can be referred back, in a manner similar to the above, to pyrone, penthiophene and pyridine, the substances containing a ring of five carbon atoms, and an oxygen, sulphur and nitrogen atom respectively. As before, only true ring nuclei, and not internal anhydrides of aliphatic compounds, will be mentioned. From the pyrone ring the following series of compounds are derived (for brevity, the hydrogen atoms are not printed):
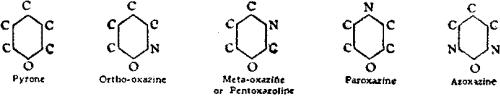
Penthiophene gives, by a similar introduction of nitrogen atoms, penthiazoline, corresponding to meta-oxazine, and para-thiazine, corresponding to paroxazine (para-oxazine). Pyridine gives origin to: pyridazine or ortho-diazine, pyrimidine or meta-diazine, pyrazine or para-diazine, osotriazine, unsymmetrical triazine, symmetrical triazine, osotetrazone and tetrazine. The skeletons of these types are (the carbon atoms are omitted for brevity):
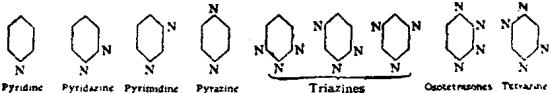
We have previously referred to the condensation of heterocyclic ring systems containing two vicinal carbon atoms with benzene, naphthalene and other nuclei. The more important nuclei of this type have received special and non-systematic names; when this is not the case, such terms as phen-, benzo-, naphtho- are prefixed to the name of the heterocyclic ring. One or two benzene nuclei may suffer condensation with the furfurane, thiophene and pyrrol rings, the common carbon atoms being vicinal to the hetero-atom. The mono-benzo-derivatives are coumarone, benzothiophene and indole; the dibenzo-derivatives are diphenylene oxide, dibenzothiophene or diphenylene sulphide, and carbazole. Typical formulae are (R denoting O, S or NH):
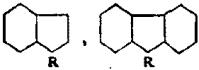
Isomers are possible, for the condensation may be effected on the two carbon atoms symmetrically placed to the hetero-atom; these isomers, however, are more of the nature of internal anhydrides. Benz-oxazoles and -thiazoles have been prepared, benz-isoxazoles are known as indoxazenes; benzo-pyrazoles occur in two structural forms, named indazoles and isindazoles. Derivatives of osotriazol also exist in two forms—azimides and pseudo-azimides.
Proceeding to the six-membered hetero-atomic rings, the benzo-, dibenzo- and naphtho-derivatives are frequently of great commercial and scientific importance, α-pyrone condenses with the benzene ring to form coumarin and isocoumarin; benzo-γ-pyrone constitutes the nucleus of several vegetable colouring matters (chrysin, fisetin, quercetin, &c., which are derivatives of flavone or phenyl benzo-γ-pyrone); dibenzo-γ-pyrone is known as xanthone; related to this substance are fluorane (and fluorescein), fluorone, fluorime, pyronine, &c. The pyridine ring condenses with the benzene ring to form quinoline and isoquinoline; acridine and phenanthridine are dibenzo-pyridines; naphthalene gives rise to α- and β-naphthoquinolines and the anthrapyridines; anthracene gives anthraquinoline; while two pyridine nuclei connected by an intermediate benzene nucleus give the phenanthrolines. Naphthyridines and naphthinolines result from the condensation of two pryridine and two quinoline nuclei respectively; and quino-quinolines are unsymmetrical naphthyridine nuclei condensed with a benzene nucleus. Benzo-orthoxazines, -metoxazines and -paroxazines are known: dibenzoparoxazine or phenoxazine is the parent of a valuable series of dyestuffs; dibenzoparathiazine or thiodiphenylamine is important from the same aspect. Benzo-ortho-diazines exist in two structural forms, cinnolin and phthalazine; benzo-meta-diazines are known as quinazolines; benzo-para-diazines are termed quinoxalines; the dibenzo-compounds are named phenazines, this last group including many valuable dyestuffs—indulines, safranines, &c. In addition to the types of compounds enumerated above we may also notice purin, tropine and the terpenes.
V. ANALYTICAL CHEMISTRY
This branch of chemistry has for its province the determination of the constituents of a chemical compound or of a mixture of compounds. Such a determination is qualitative, the constituent being only detected or proved to be present, or quantitative, in which the amount present is ascertained. The methods of chemical analysis may be classified according to the type of reaction: (1) dry or blowpipe analysis, which consists in an examination of the substance in the dry condition; this includes such tests as ignition in a tube, ignition on charcoal in the blowpipe flame, fusion with borax, microcosmic salt or fluxes, and flame colorations (in quantitative work the dry methods are sometimes termed “dry assaying”); (2) wet analysis, in which a solution of the substance is treated with reagents which produce specific reactions when certain elements or groups of elements are present. In quantitative analysis the methods can be subdivided into: (a) gravimetric, in which the constituent is precipitated either as a definite insoluble compound by the addition of certain reagents, or electrolytically, by the passage of an electric current; (b) volumetric, in which the volume of a reagent of a known strength which produces a certain definite reaction is measured; (c) colorimetric, in which the solution has a particular tint, which can be compared with solutions of known strengths.
Historical.—The germs of analytical chemistry are to be found in the writings of the pharmacists and chemists of the iatrochemical period. The importance of ascertaining the proximate composition of bodies was clearly realized by Otto Tachenius; but the first systematic investigator was Robert Boyle, to whom we owe the introduction of the term analysis. Boyle recognized many reagents which gave precipitates with certain solutions: he detected sulphuric and hydrochloric acids by the white precipitates formed with calcium chloride and silver nitrate respectively; ammonia by the white cloud formed with the vapours of nitric or hydrochloric acids; and copper by the deep blue solution formed by a solution of ammonia. Of great importance is his introduction of vegetable juices (the so-called indicators, q.v.) to detect acids and bases. During the phlogistic period, the detection of the constituents of compounds was considerably developed. Of the principal workers in this field we may notice Friedrich Hoffmann, Andreas Sigismund Marggraf (who detected iron by its reaction with potassium ferrocyanide, and potassium and sodium by their flame colorations), and especially Carl Scheele and Torbern Olof Bergman. Scheele enriched the knowledge of chemistry by an immense number of facts, but he did not possess the spirit of working systematically as Bergman did. Bergman laid the foundations of systematic qualitative analysis, and devised methods by which the metals may be separated into groups according to their behaviour with certain reagents. This subdivision, which is of paramount importance in the analysis of minerals, was subsequently developed by Wilhelm August Lampadius in his Handbuch zur chemischen Analyse der Mineralien (1801) and by John Friedrich A. Göttling in his Praktische Anleitung zur prüfenden und zurlegenden Chemie (1802).
The introduction of the blowpipe into dry qualitative analysis by Axel Fredrik Cronstedt marks an important innovation. The rapidity of the method, and the accurate results which it gave in the hands of a practised experimenter, led to its systematization by Jöns Jakob Berzelius and Johann Friedrich Ludwig Hausmann, and in more recent times by K.F. Plattner, whose treatise Die Probirkunst mit dem Löthrohr is a standard work on the subject. Another type of dry reaction, namely, the flame coloration, had been the subject of isolated notices, as, for example, the violet flame of potassium and the orange flame of sodium observed by Marggraf and Scheele, but a systematic account was wanting until Cartmell took the subject up. His results (Phil. Mag. 16, p. 382) were afterwards perfected by Robert Wilhelm Bunsen and Gustav Merz. Closely related to the flame-colorations, we have to notice the great services rendered by the spectroscope to the detection of elements. Rubidium, caesium, thallium, indium and gallium were first discovered by means of this instrument; the study of the rare earths is greatly facilitated, and the composition of the heavenly bodies alone determinable by it.
Quantitative chemistry had been all but neglected before the time of Lavoisier, for although a few chemists such as Tachenius, Bergman and others had realized the advantages which would accrue from a knowledge of the composition of bodies by weight, and had laid down the lines upon which such determinations should proceed, the experimental difficulties in making accurate observations were enormous, and little progress could be made until the procedure was more accurately determined. Martin Heinrich Klaproth showed the necessity for igniting precipitates before weighing them, if they were not decomposed by this process; and he worked largely with Louis Nicolas Vauquelin in perfecting the analysis of minerals. K.F. Wenzel and J.B. Richter contributed to the knowledge of the quantitative composition of salts. Anton Laurent Lavoisier, however, must be considered as the first great exponent of this branch of chemistry. He realized that the composition by weight of chemical compounds was of the greatest moment if chemistry were to advance. His fame rests upon his exposition of the principles necessary to chemistry as a science, but of his contributions to analytical inorganic chemistry little can be said. He applied himself more particularly to the oxygen compounds, and determined with a fair degree of accuracy the ratio of carbon to oxygen in carbon dioxide, but his values for the ratio of hydrogen to oxygen in water, and of phosphorus to oxygen in phosphoric acid, are only approximate; he introduced no new methods either for the estimation or separation of the metals. The next advance was made by Joseph Louis Proust, whose investigations led to a clear grasp of the law of constant proportions. The formulation of the atomic theory by John Dalton gave a fresh impetus to the development of quantitative analysis; and the determination of combining or equivalent weights by Berzelius led to the perfecting of the methods of gravimetric analysis. Experimental conditions were thoroughly worked out; the necessity of working with hot or cold solutions was clearly emphasized; and the employment of small quantities of substances instead of the large amounts recommended by Klaproth was shown by him to give more consistent results.
Since the time of Berzelius many experimenters have entered the lists, and introduced developments which we have not space to mention. We may, however, notice Heinrich Rose15 and Friedrich Wohler,16 who, having worked up the results of their teacher Berzelius, and combined them with their own valuable observations, exerted great influence on the progress of analytical chemistry by publishing works which contained admirable accounts of the then known methods of analysis. To K.R. Fresenius, the founder of the Zeitschrift für analytische Chemie (1862), we are particularly indebted for perfecting and systematizing the various methods of analytical chemistry. By strengthening the older methods, and devising new ones, he exerted an influence which can never be overestimated. His text-books on the subject, of which the Qualitative appeared in 1841, and the Quantitative in 1846, have a world-wide reputation, and have passed through several editions.
The quantitative precipitation of metals by the electric current, although known to Michael Faraday, was not applied to analytical chemistry until O. Wolcott Gibbs worked out the electrolytic separation of copper in 1865. Since then the subject has been extensively studied, more particularly by Alexander Classen, who has summarized the methods and results in his Quantitative Chemical Analysis by Electrolysis (1903). The ever-increasing importance of the electric current in metallurgy and chemical manufactures is making this method of great importance, and in some cases it has partially, if not wholly, superseded the older methods.
Volumetric analysis, possessing as it does many advantages over the gravimetric methods, has of late years been extensively developed. Gay Lussac may be regarded as the founder of the method, although rough applications had been previously made by F.A.H. Descroizilles and L.N. Vauquelin. Chlorimetry (1824), alkalimetry (1828), and the volumetric determination of silver and chlorine (1832) were worked out by Gay Lussac; but although the advantages of the method were patent, it received recognition very slowly. The application of potassium permanganate to the estimation of iron by E. Margueritte in 1846, and of iodine and sulphurous acid to the estimation of copper and many other substances by Robert Wilhelm Bunsen, marks an epoch in the early history of volumetric analysis. Since then it has been rapidly developed, particularly by Karl Friedrich Mohr and J. Volhard, and these methods rank side by side in value with the older and more tedious gravimetric methods.
The detection of carbon and hydrogen in organic compounds by the formation of carbon dioxide and water when they are burned was first correctly understood by Lavoisier, and as he had determined the carbon and hydrogen content of these two substances he was able to devise methods by which carbon and hydrogen in organic compounds could be estimated. In his earlier experiments he burned the substance in a known volume of oxygen, and by measuring the residual gas determined the carbon and hydrogen. For substances of a difficultly combustible nature he adopted the method in common use to-day, viz. to mix the substance with an oxidizing agent—mercuric oxide, lead dioxide, and afterwards copper oxide—and absorb the carbon dioxide in potash solution. This method has been improved, especially by Justus v. Liebig; and certain others based on a different procedure have been suggested. The estimation of nitrogen was first worked out in 1830 by Jean Baptiste Dumas, and different processes have been proposed by Will and F. Varrentrapp, J. Kjeldahl and others. Methods for the estimation of the halogens and sulphur were worked out by L. Carius (see below, § Organic Analysis).
Only a reference can be made in this summary to the many fields in which analytical chemistry has been developed. Progress in forensic chemistry was only possible after the reactions of poisons had been systematized; a subject which has been worked out by many investigators, of whom we notice K.R. Fresenius, J. and R. Otto, and J.S. Stas. Industrial chemistry makes many claims upon the chemist, for it is necessary to determine the purity of a product before it can be valued. This has led to the estimation of sugar by means of the polarimeter, and of the calorific power of fuels, and the valuation of ores and metals, of coal-tar dyes, and almost all trade products.
The passing of the Food and Drug Acts (1875-1899) in England, and the existence of similar adulteration acts in other countries, have occasioned great progress in the analysis of foods, drugs, &c. For further information on this branch of analytical chemistry, see Adulteration.
There exists no branch of technical chemistry, hygiene or pharmacy from which the analytical chemist can be spared, since it is only by a continual development of his art that we can hope to be certain of the purity of any preparation. In England this branch of chemistry is especially cared for by the Institute of Chemistry, which, since its foundation in 1877, has done much for the training of analytical chemists.
In the preceding sketch we have given a necessarily brief account of the historical development of analytical chemistry in its main branches. We shall now treat the different methods in more detail. It must be mentioned here that the reactions of any particular substance are given under its own heading, and in this article we shall only collate the various operations and outline the general procedure. The limits of space prevent any systematic account of the separation of the rare metals, the alkaloids, and other classes of organic compounds, but sources where these matters may be found are given in the list of references.
Qualitative Inorganic Analysis.
The dry examination of a substance comprises several operations, which may yield definite results if no disturbing element is present; but it is imperative that any inference Dry methods. should be confirmed by other methods.
1. Heat the substance in a hard glass tube. Note whether any moisture condenses on the cooler parts of the tube, a gas is evolved, a sublimate formed, or the substance changes colour.
Moisture is evolved from substances containing water of crystallization or decomposed hydrates. If it possesses an alkaline or acid reaction, it must be tested in the first case for ammonia, and in the second case for a volatile acid, such as sulphuric, nitric, hydrochloric, &c.
Any evolved gas must be examined. Oxygen, recognized by its power of igniting a glowing splinter, results from the decomposition of oxides of the noble metals, peroxides, chlorates, nitrates and other highly oxygenized salts. Sulphur dioxide, recognized by its smell and acid reaction, results from the ignition of certain sulphites, sulphates, or a mixture of a sulphate with a sulphide. Nitrogen oxides, recognized by their odour and brown-red colour, result from the decomposition of nitrates. Carbon dioxide, recognized by turning lime-water milky, indicates decomposable carbonates or oxalates. Chlorine, bromine, and iodine, each recognizable by its colour and odour, result from decomposable haloids; iodine forms also a black sublimate. Cyanogen and hydrocyanic acid, recognizable by their odour, indicate decomposable cyanides. Sulphuretted hydrogen, recognized by its odour, results from sulphides containing water, and hydrosulphides. Ammonia, recognizable by its odour and alkaline reaction, indicates ammoniacal salts or cyanides containing water.A sublimate may be formed of: sulphur—reddish-brown drops, cooling to a yellow to brown solid, from sulphides or mixtures; iodine—violet vapour, black sublimate, from iodides, iodic acid, or mixtures; mercury and its compounds—metallic mercury forms minute globules, mercuric sulphide is black and becomes red on rubbing, mercuric chloride fuses before subliming, mercurous chloride does not fuse, mercuric iodide gives a yellow sublimate; arsenic and its compounds—metallic arsenic gives a grey mirror, arsenious oxide forms white shining crystals, arsenic sulphides give reddish-yellow sublimates which turn yellow on cooling; antimony oxide fuses and gives a yellow acicular sublimate; lead chloride forms a white sublimate after long and intense heating.
If the substance does not melt but changes colour, we may have present: zinc oxide—from white to yellow, becoming white on cooling; stannic oxide—white to yellowish brown, dirty white on cooling; lead oxide—from white or yellowish-red to brownish-red, yellow on cooling; bismuth oxide—from white or pale yellow to orange-yellow or reddish-brown, pale yellow on cooling; manganese oxide—from white or yellowish white to dark brown, remaining dark brown on cooling (if it changes on cooling to a bright reddish-brown, it indicates cadmium oxide); copper oxide—from bright blue or green to black; ferrous oxide—from greyish-white to black; ferric oxide—from brownish-red to black, brownish-red on cooling; potassium chromate—yellow to dark orange, fusing at a red heat.
2. Heat the substance on a piece of charcoal in the reducing flame of the blowpipe.
(α) The substance may fuse and be absorbed by the charcoal; this indicates more particularly the alkaline metals.
(β) An infusible white residue may be obtained, which may denote barium, strontium, calcium, magnesium, aluminium or zinc. The first three give characteristic flame colorations (see below); the last three, when moistened with cobalt nitrate and re-ignited, give coloured masses; aluminium (or silica) gives a brilliant blue; zinc gives a green; whilst magnesium phosphates or arsenate (and to a less degree the phosphates of the alkaline earths) give a violet mass.
A metallic globule with or without an incrustation may be obtained. Gold and copper salts give a metallic bead without an incrustation. If the incrustation be white and readily volatile, arsenic is present, if more difficultly volatile and beads are present, antimony; zinc gives an incrustation yellow whilst hot, white on cooling, and volatilized with difficulty; tin gives a pale yellow incrustation, which becomes white on cooling, and does not volatilize in either the reducing or oxidizing flames; lead gives a lemon-yellow incrustation turning sulphur-yellow on cooling, together with metallic malleable beads; bismuth gives metallic globules and a dark orange-yellow incrustation, which becomes lemon-yellow on cooling; cadmium gives a reddish-brown incrustation, which is removed without leaving a gleam by heating in the reducing flame; silver gives white metallic globules and a dark-red incrustation.
3. Heat the substance with a bead of microcosmic salt or borax on a platinum wire in the oxidizing flame.
(α) The substance dissolves readily and in quantity, forming a bead which is clear when hot. If the bead is coloured we may have present: cobalt, blue to violet; copper, green, blue on cooling; in the reducing flame, red when cold; chromium, green, unaltered in the reducing flame; iron, brownish-red, light-yellow or colourless on cooling; in the reducing flame, red while hot, yellow on cooling, greenish when cold; nickel, reddish to brownish-red, yellow to reddish-yellow or colourless on cooling, unaltered in the reducing flame; bismuth, yellowish-brown, light-yellow or colourless on cooling; in the reducing flame, almost colourless, blackish-grey when cold; silver, light yellowish to opal, somewhat opaque when cold; whitish-grey in the reducing flame; manganese, amethyst red, colourless in the reducing flame. If the hot bead is colourless and remains clear on cooling, we may suspect the presence of antimony, aluminium, zinc, cadmium, lead, calcium and magnesium. When present in sufficient quantity the five last-named give enamel-white beads; lead oxide in excess gives a yellowish bead. If the hot colourless bead becomes enamel-white on cooling even when minute quantities of the substances are employed, we may infer the presence of barium or strontium.
(β) The substance dissolves slowly and in small quantity, and forms a colourless bead which remains so on cooling. Either silica or tin may be present. If silica be present, it gives the iron bead when heated with a little ferric oxide; if tin is present there is no change. Certain substances, such as the precious metals, are quite insoluble in the bead, but float about in it.
4. Hold a small portion of the substance moistened with hydrochloric acid on a clean platinum wire in the fusion zone of the Bunsen burner, and note any colour imparted to the flame.
Potassium gives a blue-violet flame which may be masked by the colorations due to sodium, calcium and other elements. By viewing the flame through an indigo prism it appears sky-blue, violet and ultimately crimson, as the thickness of the prism is increased. Other elements do not interfere with this method. Sodium gives an intense and persistent yellow flame; lithium gives a carmine coloration, and may be identified in the presence of sodium by viewing through a cobalt glass or indigo prism; from potassium it may be distinguished by its redder colour; barium gives a yellowish-green flame, which appears bluish-green when viewed through green glass; strontium gives a crimson flame which appears purple or rose when viewed through blue glass; calcium gives an orange-red colour which appears finch-green through green glass; indium gives a characteristic bluish-violet flame; copper gives an intense emerald-green coloration.
5. Film Reactions.—These reactions are practised in the following manner:—A thread of asbestos is moistened and then dipped in the substance to be tested; it is then placed in the luminous point of the Bunsen flame, and a small porcelain basin containing cold water placed immediately over the asbestos. The formation of a film is noted. The operation is repeated with the thread in the oxidizing flame.
Any film formed in the first case is metallic, in the second it is the oxide. The metallic film is tested with 20% nitric acid and with bleaching-powder solution. Arsenic is insoluble in the acid, but immediately dissolves in the bleaching-powder. The black films of antimony and bismuth and the grey mottled film of mercury are slowly soluble in the acid, and untouched by bleaching-powder. The black films of tin, lead and cadmium dissolve at once in the acid, the lead film being also soluble in bleaching-powder. The oxide films of antimony, arsenic, tin and bismuth are white, that of bismuth slightly yellowish; lead yields a very pale yellow film, and cadmium a brown one; mercury yields no oxide film. The oxide films (the metallic one in the case of mercury) are tested with hydriodic acid, and with ammonium sulphide, and from the changes produced the film can be determined (see F.M. Perkin, Qualitative Chemical Analysis, 1905).
Having completed the dry analysis we may now pass on to the wet and more accurate investigation. It is first necessary to get the substance into solution. Small portions Wet methods. should be successively tested with water, dilute hydrochloric acid, dilute nitric acid, strong hydrochloric acid, and a mixture of hydrochloric and nitric acids, first in the cold and then with warming. Certain substances are insoluble in all these reagents, and other methods, such as the fusion with sodium carbonate and potassium nitrate, and subsequent treatment with an acid, must be employed. Some of these insoluble compounds can be detected by their colour and particular reactions. For further information on this subject, we refer the readers to Fresenius’s Qualitative Analysis.
The procedure for the detection of metals in solution consists of first separating them into groups and then examining each group separately. For this purpose the cold solution is treated with hydrochloric acid, which precipitates lead, silver and mercurous salts as chlorides. The solution is filtered and treated with an excess of sulphuretted hydrogen, either in solution or by passing in the gas; this precipitates mercury (mercuric), any lead left over from the first group, copper, bismuth, cadmium, arsenic, antimony and tin as sulphides. The solution is filtered off, boiled till free of sulphuretted hydrogen, and ammonium chloride and ammonia added. If phosphoric acid is absent, aluminium, chromium and ferric hydrates are precipitated. If, however, phosphoric acid is present in the original substance, we may here obtain a precipitate of the phosphates of the remaining metals, together with aluminium, chromium and ferric hydrates. In this case, the precipitate is dissolved in as little as possible hydrochloric acid and boiled with ammonium acetate, acetic acid and ferric chloride. The phosphates of aluminium, chromium and iron are precipitated, and the solution contains the same metals as if phosphoric acid had been absent. To the filtrate from the aluminium, iron and chromium precipitate, ammonia and ammonium sulphide are added; the precipitate may contain nickel, cobalt, zinc and manganese sulphides. Ammonium carbonate is added to the filtrate; this precipitates calcium, strontium and barium. The solution contains magnesium, sodium and potassium, which are separately distinguished by the methods given under their own headings.
We now proceed with the examination of the various group precipitates. The white precipitate formed by cold hydrochloric acid is boiled with water, and the solution filtered while hot. Any lead chloride dissolves, and may be identified by the yellow precipitate formed with potassium chromate. To the residue add ammonia, shake, then filter. Silver chloride goes into solution, and may be precipitated by dilute nitric acid. The residue, which is black in colour, consists of mercuroso-ammonium chloride, in which mercury can be confirmed by its ordinary tests.
The precipitate formed by sulphuretted hydrogen may contain the black mercuric, lead, and copper sulphides, dark-brown bismuth sulphide, yellow cadmium and arsenious sulphides, orange-red antimony sulphide, brown stannous sulphide, dull-yellow stannic sulphide, and whitish sulphur, the last resulting from the oxidation of sulphuretted hydrogen by ferric salts, chromates, &c. Warming with ammonium sulphide dissolves out the arsenic, antimony and tin salts, which are reprecipitated by the addition of hydrochloric acid to the ammonium sulphide solution. The precipitate is shaken with ammonium carbonate, which dissolves the arsenic. Filter and confirm arsenic in the solution by its particular tests. Dissolve the residue in hydrochloric acid and test separately for antimony and tin. The residue from the ammonium sulphide solution is warmed with dilute nitric acid. Any residue consists of black mercuric sulphide (and possibly white lead sulphate), in which mercury is confirmed by its usual tests. The solution is evaporated with a little sulphuric acid and well cooled. The white precipitate consists of lead sulphate. To the filtrate add ammonia in excess; a white precipitate indicates bismuth; if the solution be blue, copper is present. Filter from the bismuth hydrate, and if copper is present, add potassium cyanide till the colour is destroyed, then pass sulphuretted hydrogen, and cadmium is precipitated as the yellow sulphide. If copper is absent, then sulphuretted hydrogen can be passed directly into the solution.
The next group precipitate may contain the white gelatinous aluminium hydroxide, the greenish chromium hydroxide, reddish ferric hydroxide, and possibly zinc and manganese hydroxides. Treatment with casutic soda dissolves out aluminium hydroxide, which is reprecipitated by the addition of ammonium chloride. The remaining metals are tested for separately.
The next group may contain black nickel and cobalt sulphides, flesh-coloured manganese sulphide, and white zinc sulphide. The last two are dissolved out by cold, very dilute hydrochloric acid, and the residue is tested for nickel and cobalt. The solution is boiled till free from sulphuretted hydrogen and treated with excess of sodium hydrate. A white precipitate rapidly turning brown indicates manganese. The solution with ammonium sulphide gives a white precipitate of zinc sulphide.
The next group may contain the white calcium, barium and strontium carbonates. The flame coloration (see above) may give information as to which elements are present. The carbonates are dissolved in hydrochloric acid, and calcium sulphate solution is added to a portion of the solution. An immediate precipitate indicates barium; a precipitate on standing indicates strontium. If barium is present, the solution of the carbonates in hydrochloric acid is evaporated and digested with strong alcohol for some time; barium chloride, which is nearly insoluble in alcohol, is thus separated, the remainder being precipitated by a few drops of hydrofluosilicic acid, and may be confirmed by the ordinary tests. The solution free from barium is treated with ammonia and ammonium sulphate, which precipitates strontium, and the calcium in the solution may be identified by the white precipitate with ammonium oxalate.
Having determined the bases, it remains to determine the acid radicals. There is no general procedure for these operations, and it is customary to test for the acids separately by special tests; these are given in the articles on the various acids. A knowledge of the solubility of salts considerably reduces the number of acids likely to be present, and affords evidence of great value to the analyst (see A.M. Comey, Dictionary of Chemical Solubilities.) In the above account we have indicated the procedure adopted in the analysis of a complex mixture of salts. It is unnecessary here to dwell on the precautions which can only be conveniently acquired by experience; a sound appreciation of analytical methods is only possible after the reactions and characters of individual substances have been studied, and we therefore refer the reader to the articles on the particular elements and compounds for more information on this subject.
Quantitative Inorganic Analysis.
Quantitative methods are divided into four groups, which we now pass on to consider in the following sequence: (α) gravimetric, (β) volumetric, (γ) electrolytic, (δ) colorimetric.
(α) Gravimetric.—This method is made up of four operations: (1) a weighed quantity of the substance is dissolved in a suitable solvent; (2) a particular reagent is added which precipitates the substance it is desired to estimate; (3) the precipitate is filtered, washed and dried; (4) the filter paper containing the precipitate is weighed either as a tared filter, or incinerated and ignited either in air or in any other gas, and then weighed.
(1) Accurate weighing is all-important: for details of the various appliances and methods see Weighing Machines. (2) No general directions can be given as to the method of precipitation. Sometimes it is necessary to allow the solution to stand for a considerable time either in the warm or cold or in the light or dark; to work with cold solutions and then boil; or to use boiling solutions of both the substance and reagent. Details will be found in the articles on particular metals. (3) The operation of filtration and washing is very important. If the substance to be weighed changes in composition on strong heating, it is necessary to employ a tared filter, i.e. a filter paper which has been previously heated to the temperature at which the substance is to be dried until its weight is constant. If the precipitate settles readily, the supernatant liquor may be decanted through the filter paper, more water added to the precipitate and again decanted. By this means most of the washing, i.e.freeing from the other substances in the solution, can be accomplished in the precipitating vessel. If, however, the precipitate refuses to settle, it is directly transferred to the filter paper, the last traces being removed by washing and rubbing the sides of the vessel with a piece of rubber, and the liquid is allowed to drain through. It is washed by ejecting a jet of water, ammonia or other prescribed liquid on to the side of the filter paper until the paper is nearly full. It can be shown that a more efficient washing results from alternately filling and emptying the funnel than by endeavouring to keep the funnel full. The washing is continued until the filtrate is free from salts or acids. (4) After washing, the funnel containing the filter paper is transferred to a drying oven. In the case of a tared filter it is weighed repeatedly until the weight suffers no change; then knowing the weight of the filter paper, the weight of the precipitate is obtained by subtraction. If the precipitate may be ignited, it is transferred to a clean, weighed and recently ignited crucible, and the filter paper is burned separately on the lid, the ash transferred to the crucible, and the whole ignited. After ignition, it is allowed to cool in a desiccator and then weighed. Knowing the weight of the crucible and of the ash of the filter paper, the weight of the precipitate is determined. The calculation of the percentage of the particular constituent is simple. We know the amount present in the precipitate, and since the same amount is present in the quantity of substance experimented with, we have only to work out a sum in proportion.
(β) Volumetric.—This method is made up of three operations:—(1) preparation of a standard solution; (2) preparation of a solution of the substance; (3) titration, or the determination of what volume of the standard solution will occasion a known and definite reaction with a known volume of the test solution.
(1) In general analytical work the standard solution contains the equivalent weight of the substance in grammes dissolved in a litre of water. Such a solution is known as normal. Thus a normal solution of sodium carbonate contains 53 grammes per litre, of sodium hydrate 40 grammes, of hydrochloric acid 36.5 grammes, and so on. By taking 1/10th or 1/100th of these quantities, decinormal or centinormal solutions are obtained. We see therefore that 1 cubic centimetre of a normal sodium carbonate solution will exactly neutralize 0.049 gramme of sulphuric acid, 0.0365 gramme of hydrochloric acid (i.e. the equivalent quantities), and similarly for decinormal and centinormal solutions. Unfortunately, the term normal is sometimes given to solutions which are strictly decinormal; for example, iodine, sodium thiosulphate, &c. In technical analysis, where a solution is used for one process only, it may be prepared so that 1 cc. is equal to .01 gramme of the substance to be estimated. This saves a certain amount of arithmetic, but when the solution is applied in another determination additional calculations are necessary. Standard solutions are prepared by weighing out the exact amount of the pure substance and dissolving it in water, or by forming a solution of approximate normality, determining its exact strength by gravimetric or other means, and then correcting it for any divergence. This may be exemplified in the case of alkalimetry. Pure sodium carbonate is prepared by igniting the bicarbonate, and exactly 53 grammes are dissolved in water, forming a strictly normal solution. An approximate normal sulphuric acid is prepared from 30 ccs. of the pure acid (1.84 specific gravity) diluted to 1 litre. The solutions are titrated (see below) and the acid solution diluted until equal volumes are exactly equivalent. A standard sodium hydrate solution can be prepared by dissolving 42 grammes of sodium hydrate, making up to a litre, and diluting until one cubic centimetre is exactly equivalent to one cubic centimetre of the sulphuric acid. Similarly, normal solutions of hydrochloric and nitric acids can be prepared. Where a solution is likely to change in composition on keeping, such as potassium permanganate, iodine, sodium hydrate, &c., it is necessary to check or re-standardize it periodically.
(2) The preparation of the solution of the substance consists in dissolving an accurately determined weight, and making up the volume in a graduated cylinder or flask to a known volume.
(3) The titration is conducted by running the standard solution from a burette into a known volume of the test solution, which is usually transferred from the stock-bottle to a beaker or basin by means of a pipette. Various artifices are employed to denote the end of the reaction. These may be divided into two groups: (1) those in which a change in appearance of the reacting mixture occurs; (2) those in which it is necessary to use an indicator which, by its change in appearance, shows that an excess of one reagent is present. In the first group, we have to notice the titration of a cyanide with silver nitrate, when a milkiness shows how far the reaction has gone; the titration of iron with permanganate, when the faint pink colour shows that all the iron is oxidized. In the second group, we may notice the application of litmus, methyl orange or phenolphthalein in alkalimetry, when the acid or alkaline character of the solution commands the colour which it exhibits; starch paste, which forms a blue compound with free iodine in iodometry; potassium chromate, which forms red silver chromate after all the hydrochloric acid is precipitated in solutions of chlorides; and in the estimation of ferric compounds by potassium bichromate, the indicator, potassium ferricyanide, is placed in drops on a porcelain plate, and the end of the reaction is shown by the absence of a blue coloration when a drop of the test solution is brought into contact with it.
(γ) Electrolytic.—This method consists in decomposing a solution of a salt of the metal by the electric current and weighing the metal deposited at the cathode.
It is only by paying great attention to the current density that good results are obtained, since metals other than that sought for may be deposited. In acid copper solutions, mercury is deposited before the copper with which it subsequently amalgamates; silver is thrown down simultaneously; bismuth appears towards the end; and after all the copper has been precipitated, arsenic and antimony may be deposited. Lead and manganese are partially separated as peroxides, but the remaining metals are not deposited from acid solutions. It is therefore necessary that the solution should be free from metals which may vitiate the results, or special precautions taken by which the impurities are rendered harmless. In such cases the simplicity of manipulation and the high degree of accuracy of the method have made it especially valuable. The electrolysis is generally conducted with platinum electrodes, of which the cathode takes the form of a piece of foil bent into a cylindrical form, the necessary current being generated by one or more Daniell cells.
(δ) Colorimetric.—This method is adopted when it is necessary to determine minute traces (as in the liquid obtained in the electrolytic separation of copper) of substances which afford well-defined colour reactions.
The general procedure is to make a series of standard solutions containing definite quantities of the substance which it is desired to estimate; such a series will exhibit tints which deepen as the quantity of the substance is increased. A known weight of the test substance is dissolved and a portion of the solution is placed in a tube similar to those containing the standard solutions. The colour-producing reagent is added and the tints compared. In the case of copper, the colour reactions with potassium ferrocyanide or ammonia are usually employed; traces of ammonia are estimated with Nessler’s reagent; sulphur in iron and steel is determined by the tint assumed by a silver-copper plate suspended in the gases liberated when the metal is dissolved in sulphuric acid (Eggertz’s test) (see W. Crookes, Select Methods in Analytical Chemistry).
Organic Analysis.
The elements which play important parts in organic compounds are carbon, hydrogen, nitrogen, chlorine, bromine, iodine, sulphur, phosphorus and oxygen. We shall here consider the qualitative and quantitative determination of these elements.
Qualitative.—Carbon is detected by the formation of carbon dioxide, which turns lime-water milky, and hydrogen by the formation of water, which condenses on the tube, when the substance is heated with copper oxide. Nitrogen may be detected by the evolution of ammonia when the substance is heated with soda-lime. A more delicate method is that due to J. L. Lassaigne and improved by O. Jacobsen and C. Graebe. The substance is heated with metallic sodium or potassium (in excess if sulphur be present) to redness, the residue treated with water, filtered, and ferrous sulphate, ferric chloride and hydrochloric acid added. A blue coloration indicates nitrogen, and is due to the formation of potassium (or sodium) cyanide during the fusion, and subsequent interaction with the iron salts. The halogens may be sometimes detected by fusing with lime, and testing the solution for a bromide, chloride and iodide in the usual way. F. Beilstein determines their presence by heating the substance with pure copper oxide on a platinum wire in the Bunsen flame; a green coloration is observed if halogens be present. Sulphur is detected by heating the substance with sodium, dissolving the product in water, and adding sodium nitroprusside; a bluish-violet coloration indicates sulphur (H. Vohl). Or we may use J. Horbaczewski’s method, which consists in boiling the substance with strong potash, saturating the cold solution with chlorine, adding hydrochloric acid, and boiling till no more chlorine is liberated, and then testing for sulphuric acid with barium chloride. Phosphorus is obtained as a soluble phosphate (which can be examined in the usual way) by lixiviating the product obtained when the substance is ignited with potassium nitrate and carbonate.
Quantitative.—Carbon and hydrogen are generally estimated by the combustion process, which consists in oxidizing the substance and absorbing the products of combustion in suitable Carbon and hydrogen. apparatus. The oxidizing agent in commonest use is copper oxide, which must be freshly ignited before use on account of its hygroscopic nature. Lead chromate is sometimes used, and many other substances, such as platinum, manganese dioxide, &c., have been suggested. The procedure for a combustion is as follows:—
A hard glass tube slightly longer than the furnace and 12 to 15 mm. in diameter is thoroughly cleansed and packed as shown in fig. 1. The space a must allow for the inclusion of a copper spiral if the substance contains nitrogen, and a silver spiral if halogens be present, for otherwise nitrogen oxides and the halogens may be condensed in the absorption apparatus; b contains copper oxide; c is a space for the insertion of a porcelain or platinum boat containing a weighed quantity of the substance; d is a copper spiral. The end d is connected to an air or oxygen supply with an intermediate drying apparatus. The other end is connected with the absorption vessels, which consist of a tube (e) containing calcium chloride, and a set of bulbs (f) containing potash solution. Various forms of potash bulbs are employed; fig. 2 is Liebig’s, fig. 3 Mohr’s or Geissler’s, fig. 4 is a more recent form, of which special variations have been made by Anderson, Gomberg, Delisle and others. After having previously roasted the tube and copper oxide, and reduced the copper spiral a, the weighed calcium chloride tube and potash bulbs are put in position, the boat containing the substance is inserted (in the case of a difficultly combustible substance it is desirable to mix it with cupric oxide or lead chromate), the copper spiral (d) replaced, and the air and oxygen supply connected up. The apparatus is then tested for leaks. If all the connexions are sound, the copper oxide is gradually heated from the end a, the gas-jets under the spiral d are lighted, and a slow current of oxygen is passed through the tube. The success of the operation depends upon the slow burning of the substance. Towards the end the heat and the oxygen supply are increased. When there is no more absorption in the potash bulbs, the oxygen supply is cut off and air passed through. Having replaced the oxygen in the absorption vessels by air, they are disconnected and weighed, after having cooled down to the temperature of the room. The increase in weight of the calcium chloride tube gives the weight of water formed, and of the potash bulbs the carbon dioxide.
Liquids are amenable to the same treatment, but especial care must be taken so that they volatilize slowly. Difficultly volatile liquids may be weighed directly into the boat; volatile liquids are weighed in thin hermetically sealed bulbs, the necks of which are broken just before they are placed in the combustion tube.
The length of time and other disadvantages attending the combustion method have caused investigators to devise other processes. In 1855 C. Brunner described a method for oxidizing the carbon to carbon dioxide, which could be estimated by the usual methods, by heating the substance with potassium bichromate and sulphuric acid. This process has been considerably developed by J. Messinger, and we may hope that with subsequent improvements it may be adapted to all classes of organic compounds. The oxidation, which is effected by chromic acid and sulphuric acid, is conducted in a flask provided with a funnel and escape tube, and the carbon dioxide formed is swept by a current of dry air, previously freed from carbon dioxide, through a drying tube to a set of potash bulbs and a tube containing soda-lime; if halogens are present, a small wash bottle containing potassium iodide, and a U tube containing glass wool moistened with silver nitrate on one side and strong sulphuric acid on the other, must be inserted between the flask and the drying tube. The increase in weight of the potash bulbs and soda-lime tube gives the weight of carbon dioxide evolved. C.F. Cross and E.J. Bevan collected the carbon dioxide obtained in this way over mercury. They also showed that carbon monoxide was given off towards the end of the reaction, and oxygen was not evolved unless the temperature exceeded 100°.
Methods depending upon oxidation in the presence of a contact substance have come into favour during recent years. In that of M. Dennstedt, which was first proposed in 1902, the substance is vaporized in a tube containing at one end platinum foil, platinized quartz, or platinized asbestos. The platinum is maintained at a bright red heat, either by a gas flame or by an electric furnace, and the vapour is passed over it by leading in a current of oxygen. If nitrogen be present, a boat containing dry lead peroxide and heated to 320° is inserted, the oxide decomposing any nitrogen peroxide which may be formed. The same absorbent quantitatively takes up any halogen and sulphur which may be present. The process is therefore adapted to the simultaneous estimation of carbon, hydrogen, the halogens and sulphur.
Nitrogen is estimated by (1) Dumas’ method, which consists in heating the substance with copper oxide and measuring the volume of nitrogen liberated; (2) by Will and Varrentrapp’s Sidenote: Nitrogen. method, in which the substance is heated with soda-lime, and the ammonia evolved is absorbed in hydrochloric acid, and thence precipitated as ammonium chlorplatinate or estimated volumetrically; or (3) by Kjeldahl’s method, in which the substance is dissolved in concentrated sulphuric acid, potassium permanganate added, the liquid diluted and boiled with caustic soda, and the evolved ammonia absorbed in hydrochloric acid and estimated as in Will and Varrentrapp’s method.
Dumas’ Method.—In this method the operation is carried out in a hard glass tube sealed at one end and packed as shown in fig. 5. The magnesite (a) serves for the generation of carbon dioxide which clears the tube of air before the compound (mixed with fine copper oxide (b)) is burned, and afterwards sweeps the liberated nitrogen into the receiving vessel (e), which contains a strong potash solution; c is coarse copper oxide; and d a reduced copper gauze spiral, heated in order to decompose any nitrogen oxides. Ulrich Kreusler generates the carbon dioxide in a separate apparatus, and in this case the tube is drawn out to a capillary at the end (a). This artifice is specially valuable when the substance decomposes or volatilizes in a warm current of carbon dioxide. Various forms of the absorbing apparatus (e) have been discussed by M. Ilinski (Ber. 17, p. 1347), who has also suggested the use of manganese carbonate instead of magnesite, since the change of colour enables one to follow the decomposition. Substances which burn with difficulty may be mixed with mercuric oxide in addition to copper oxide.
Will and Varrentrapp’s Method.—This method, as originally proposed, is not in common use, but has been superseded by Kjeldahl’s method, since the nitrogen generally comes out too low. It is susceptible of wider application by mixing reducing agents with the soda-lime: thus Goldberg (Ber. 16, p. 2546) uses a mixture of soda-lime, stannous chloride and sulphur for nitro- and azo-compounds, and C. Arnold (Ber. 18, p. 806) a mixture containing sodium hyposulphite and sodium formate for nitrates.
Kjeldahl’s Method.—This method rapidly came into favour on account of its simplicity, both of operation and apparatus. Various substances other than potassium permanganate have been suggested for facilitating the operation; J.W. Gunning (Z. anal. Chem., 1889, p. 189) uses potassium sulphate; Lassar-Cohn uses mercuric oxide. The applicability of the process has been examined by F.W. Dafert (Z. anal. Chem., 1888, p. 224), who has divided nitrogenous bodies into two classes with respect to it. The first class includes those substances which require no preliminary treatment, and comprises the amides and ammonium compounds, pyridines, quinolines, alkaloids, albumens and related bodies; the second class requires preliminary treatment and comprises, with few exceptions, the nitro-, nitroso-, azo-, diazo- and amidoazo-compounds, hydrazines, derivatives of nitric and nitrous acids, and probably cyanogen compounds. Other improvements have been suggested by Dyer (J.C.S. Trans. 67, p. 811). For an experimental comparison of the accuracy of the Dumas, Will-Varrentrapp and Kjeldahl processes see L. L’Hôte, C.R. 1889, p. 817. Debordeaux (C.R. 1904, p. 905) has obtained good results by distilling the substance with a mixture of potassium thiosulphate and sulphide.
The halogens may be estimated by ignition with quicklime, or by heating with nitric acid and silver nitrate in a sealed tube. In the first method the substance, mixed with quicklime free from chlorine, is heated in a tube closed at one end in a combustion furnace. Halogens, sulphur, phosphorus. The product is dissolved in water, and the calcium haloid estimated in the usual way. The same decomposition may be effected by igniting with iron, ferric oxide and sodium carbonate (E. Kopp, Ber. 10, p. 290); the operation is easier if the lime be mixed with sodium carbonate, or a mixture of sodium carbonate and potassium nitrate be used. With iodine compounds, iodic acid is likely to be formed, and hence the solution must be reduced with sulphurous acid before precipitation with silver nitrate. C. Zulkowsky (Ber. 18, R. 648) burns the substance in oxygen, conducts the gases over platinized sand, and collects the products in suitable receivers. The oxidation with nitric acid in sealed tubes at a temperature of 150° to 200° for aliphatic compounds, and 250° to 260° for aromatic compounds, is in common use, for both the sulphur and phosphorus can be estimated, the former being oxidized to sulphuric acid and the latter to phosphoric acid. This method was due to L. Carius (Ann. 136, p. 129). R. Klason (Ber. 19, p. 1910) determines sulphur and the halogens by oxidizing the substance in a current of oxygen and nitrous fumes, conducting the vapours over platinum foil, and absorbing the vapours in suitable receivers. Sulphur and phosphorus can sometimes be estimated by Messinger’s method, in which the oxidation is effected by potassium permanganate and caustic alkali, or by potassium bichromate and hydrochloric acid. A comparison of the various methods for estimating sulphur has been given by O. Hammarsten (Zeit. physiolog. Chem. 9, p. 273), and by Höland (Chemiker Zeitung, 1893, p. 991). H.H. Pringsheim (Ber. 38, p. 1434) has devised a method in which the oxidation is effected by sodium peroxide; the halogens, phosphorus and sulphur can be determined by one operation.
VI. PHYSICAL CHEMISTRY
We have seen how chemistry may be regarded as having for its province the investigation of the composition of matter, and the changes in composition which matter or energy may effect on matter, while physics is concerned with the general properties of matter. A physicist, however, does more than merely quantitatively determine specific properties of matter; he endeavours to establish mathematical laws which co-ordinate his observations, and in many cases the equations expressing such laws contain functions or terms which pertain solely to the chemical composition of matter. One example will suffice here. The limiting law expressing the behaviour of gases under varying temperature and pressure assumes the form pv = RT; so stated, this law is independent of chemical composition and may be regarded as a true physical law, just as much as the law of universal gravitation is a true law of physics. But this relation is not rigorously true; in fact, it does not accurately express the behaviour of any gas. A more accurate expression (see Condensation of Gases and Molecule) is (p + a/v²)(v - b) = RT, in which a and b are quantities which depend on the composition of the gas, and vary from one gas to another.
It may be surmised that the quantitative measures of most physical properties will be found to be connected with the chemical nature of substances. In the investigation of these relations the physicist and chemist meet on common ground; this union has been attended by fruitful and far-reaching results, and the correlation of physical properties and chemical composition is one of the most important ramifications of physical chemistry. This branch receives treatment below. Of considerable importance, also, are the properties of solids, liquids and gases in solution. This subject has occupied a dominant position in physico-chemical research since the investigations of van’t Hoff and Arrhenius. This subject is treated in the article Solution; for the properties of liquid mixtures reference should also be made to the article Distillation.
Another branch of physical chemistry has for its purpose the quantitative study of chemical action, a subject which has brought out in clear detail the analogies of chemical and physical equilibrium (see Chemical Action). Another branch, related to energetics (q.v.), is concerned with the transformation of chemical energy into other forms of energy—heat, light, electricity. Combustion is a familiar example of the transformation of chemical energy into heat and light; the quantitative measures of heat evolution or absorption (heat of combustion or combination), and the deductions therefrom, are treated in the article Thermochemistry. Photography (q.v.) is based on chemical action induced by luminous rays; apart from this practical application there are many other cases in which actinic rays occasion chemical actions; these are treated in the article Photochemistry. Transformations of electrical into chemical energy are witnessed in the processes of electrolysis (q.v.; see also Electrochemistry and Electrometallurgy). The converse is presented in the common electric cell.
Physical Properties and Composition.
For the complete determination of the chemical structure of any compound, three sets of data are necessary: (1) the empirical chemical composition of the molecule; (2) the constitution, i.e. the manner in which the atoms are linked together; and (3) the configuration of the molecule, i.e. the arrangement of the atoms in space. Identity in composition, but difference in constitution, is generally known as “isomerism” (q.v.), and compounds satisfying this relation differ in many of their physical properties. If, however, two compounds only differ with regard to the spatial arrangement of the atoms, the physical properties may be (1) for the most part identical, differences, however, being apparent with regard to the action of the molecules on polarized light, as is the case when the configuration is due to the presence of an asymmetric atom (optical isomerism); or (2) both chemical and physical properties may be different when the configuration is determined by the disposition of the atoms or groups attached to a pair of doubly-linked atoms, or to two members of a ring system (geometrical isomerism or allo-isomerism). Three sets of physical properties may therefore be looked for: (1) depending on composition, (2) depending on constitution, and (3) depending on configuration. The first set provides evidence as to the molecular weight of a substance: these are termed “colligative properties.” The second and third sets elucidate the actual structure of the molecule: these are known as “constitutional properties.”
In any attempts to gain an insight into the relations between the physical properties and chemical composition of substances, the fact must never be ignored that a comparison can only be made when the particular property under consideration is determined under strictly comparable conditions, in other words, when the molecular states of the substances experimented upon are identical. This is readily illustrated by considering the properties of gases—the simplest state of aggregation. According to the law of Avogadro, equal volumes of different gases under the same conditions of temperature and pressure contain equal numbers of molecules; therefore, since the density depends upon the number of molecules present in unit volume, it follows that for a comparison of the densities of gases, the determinations must be made under coincident conditions, or the observations reduced or re-computed for coincident conditions. When this is done, such densities are measures of the molecular weights of the substances in question.
Volume Relations.17—When dealing with colligative properties of liquids it is equally necessary to ensure comparability of conditions. In the article Condensation of Gases (see also Molecule) it is shown that the characteristic equation of gases and liquids is conveniently expressed in the form (p + a/v²)(v - b) = RT. This equation, which is mathematically deducible from the kinetic theory of gases, expresses the behaviour of gases, the phenomena of the critical state, and the behaviour of liquids; solids are not accounted for. If we denote the critical volume, pressure and temperature by Vk, Pk and Tk, then it may be shown, either by considering the characteristic equation as a perfect cube in v or by using the relations that dp/dv = 0, d²p/dv² = 0 at the critical point, that Vk = 3b, Pk = a/27b², Tk = 8a/27b. Eliminating a and b between these relations, we derive PkVk/Tk = (3/8)R, a relation which should hold between the critical constants of any substance. Experiment, however, showed that while the quotient on the left hand of this equation was fairly constant for a great number of substances, yet its value was not (3/8)R but (1/3.7)R; this means that the critical density is, as a general rule, 3.7 times the theoretical density. Deviation from this rule indicates molecular dissociation or association. By actual observations it has been shown that ether, alcohol, many esters of the normal alcohols and fatty acids, benzene, and its halogen substitution products, have critical constants agreeing with this originally empirical law, due to Sydney Young and Thomas; acetic acid behaves abnormally, pointing to associated molecules at the critical point.
The critical volume provides data which may be tested for additive relations. Theoretically the critical volume is three times the volume at absolute zero, i.e. the actual volume of the Volume at critical point and at absolute zero. molecules; this is obvious by considering the result of making T zero in the characteristic equation. Experimentally (by extrapolation from the “law of the rectilinear diameter”) the critical volume is four times the volume at absolute zero (see Condensation of Gases). The most direct manner in which to test any property for additive relations is to determine the property for a number of elements, and then investigate whether these values hold for the elements in combination. Want of data for the elements, however, restricts this method to narrow limits, and hence an indirect method is necessary. It is found that isomers have nearly the same critical volume, and that equal differences in molecular content occasion equal differences in critical volume. For example, the difference due to an increment of CH2 is about 56.6, as is shown in the following table:—
| Name. | Formula. | Crit. Vol. | Vol. per CH2 | |
| Methyl formate | H·CO2CH3 | 171 | ||
| Ethyl formate | H·C02C2H5 | 228 | }227.5 | 56.5 |
| Methyl acetate | CH3·CO2CH3 | 227 | ||
| Propyl formate | H·CO2C3H7 | 284 | }283.3 | 55.8 |
| Ethyl acetate | CH3·C02C2H5 | 285 | ||
| Methyl propionate | C2H5·CO2CH3 | 28l | ||
| Propyl acetate | CH3·CO2C3H7 | 343 | }340.7 | 57.4 |
| Ethyl propionate | C2H5·CO2C2H5 | 343 | ||
| Methyl n-butyrate | }C3H7·CO2CH3 | 339 | ||
| Methyl isobutyrate | 337 | |||
Since the critical volume of normal pentane C5H12 is 307.2, we have H2 = C5H12 - 5CH2 = 307.2 - 5 × 56.6 = 24.2, and C = CH2 - H2 = 32.4. The critical volume of oxygen can be deduced from the data of the above table, and is found to be 29, whereas the experimental value is 25.
The researches of H. Kopp, begun in 1842, on the molecular volumes, i.e. the volume occupied by one gramme molecular weight of a substance, of liquids measured at their boiling-point Volume at boiling-point. under atmospheric pressure, brought to light a series of additive relations which, in the case of carbon compounds, render it possible to predict, in some measure, the composition of the substance. In practice it is generally more convenient to determine the density, the molecular volume being then obtained by dividing the molecular weight of the substance by the density. By the indirect method Kopp derived the following atomic volumes:
| C. | O. | H. | Cl. | Br. | I. | S. |
| 11 | 12.2 | 5.5 | 22.8 | 27.8 | 37.5 | 22.6. |
These values hold fairly well when compared with the experimental values determined from other compounds, and also with the molecular volumes of the elements themselves. Thus the actually observed densities of liquid chlorine and bromine at the boiling-points are 1.56 and 2.96, leading to atomic volumes 22.7 and 26.9, which closely correspond to Kopp’s values deduced from organic compounds.
These values, however, require modification in certain cases, for discrepancies occur which can be reconciled in some cases by assuming that the atomic value of a polyvalent element varies according to the distribution of its valencies. Thus a double bond of oxygen, as in the carbonyl group CO, requires a larger volume than a single bond, as in the hydroxyl group -OH, being about 12.2 in the first case and 7.8 in the second. Similarly, an increase of volume is associated with doubly and trebly linked carbon atoms.
Recent researches have shown that the law originally proposed by Kopp—“That the specific volume of a liquid compound (molecular volume) at its boiling-point is equal to the sum of the specific volumes of its constituents (atomic volumes), and that every element has a definite atomic value in its compounds”—is by no means exact, for isomers have different specific volumes, and the volume for an increment of CH2 in different homologous series is by no means constant; for example, the difference among the esters of the fatty acids is about 57, whereas for the aliphatic aldehydes it is 49. We may therefore conclude that the molecular volume depends more upon the internal structure of the molecule than its empirical content. W. Ostwald (Lehr. der allg. Chem.), after an exhaustive review of the material at hand, concluded that simple additive relations did exist but with considerable deviations, which he ascribed to differences in structure. In this connexion we may notice W. Städel’s determinations:
| CH3CCl3 | 108 | CHClBr·CH3 | 96·5 |
| CH2Cl·CHCl2 | 102.8 | CH2Br·CH2Cl | 88 |
These differences do not disappear at the critical point, and hence the critical volumes are not strictly additive.
Theoretical considerations as to how far Kopp was justified in choosing the boiling-points under atmospheric pressure as being comparable states for different substances now claim our attention. Van der Waal’s equation (p+a/v²)(v-b) = RT contains two constants a and b determined by each particular substance. If we express the pressure, volume and temperature as fractions of the critical constants, then, calling these fractions the “reduced” pressure, volume and temperature, and denoting them by π, φ and θ respectively, the characteristic equation becomes (π+3/φ²)(3φ-1) = 8θ; which has the same form for all substances. Obviously, therefore, liquids are comparable when the pressures, volumes and temperatures are equal fractions of the critical constants. In view of the extremely slight compressibility of liquids, atmospheric pressure may be regarded as a coincident condition; also C.M. Guldberg pointed out that for the most diverse substances the absolute boiling-point is about two-thirds of the critical temperature. Hence within narrow limits Kopp’s determinations were carried out under coincident conditions, and therefore any regularities presented by the critical volumes should be revealed in the specific volumes at the boiling-point.
The connexion between the density and chemical composition of solids has not been investigated with the same completeness as in the case of gases and liquids. The relation between the atomic Volume relations of solids. volumes and the atomic weights of the solid elements exhibits the periodicity which generally characterizes the elements. The molecular volume is additive in certain cases, in particular of analogous compounds of simple constitution. For instance, constant differences are found between the chlorides, bromides and iodides of sodium and potassium:—
| I. | Diff. | II. | Diff. | Diff. I. & II. |
| KCl = 37.4 | 6.9 | NaCl = 27.1 | 6.7 | 10.3 |
| KBr = 44.3 | 9.7 | NaBr = 33.8 | 9.7 | 10.5 |
| KI = 54.0 | NaI = 43.5 | 10.5 |
According to H. Schroeder the silver salts of the fatty acids exhibit additive relations; an increase in the molecule of CH2 causes an increase in the molecular volume of about 15.3.
Thermal Relations.
Specific Heat and Composition.—-The nature and experimental determination of specific heats are discussed in the article CALORIMETRY; here will be discussed the relations existing between the heat capacities of elements and compounds.
In the article Thermodynamics it is shown that the amount of heat required to raise a given weight of a gas through a certain range of temperature is different according as the gas Specific heat of gases. is maintained at constant pressure, the volume increasing, or at constant volume, the pressure increasing. A gas, therefore, has two specific heats, generally denoted by Cp and Cv, when the quantity of gas taken as a unit is one gramme molecular weight, the range of temperature being 1° C. It may be shown that Cp - Cv = R, where R is the gas-constant, i.e. R in the equation PV = RT. From the ratio Cp/Cv conclusions may be drawn as to the molecular condition of the gas. By considerations based on the kinetic theory of gases (see Molecule) it may be shown that when no energy is utilized in separating the atoms of a molecule, this ratio is 5/3 = 1.67. If, however, an amount of energy a is taken up in separating atoms, the ratio is expressible as Cp/Cv = (5+a)/(3+a), which is obviously smaller than 5/3, and decreases with increasing values of a. These relations may be readily tested, for the ratio Cp/Cv is capable of easy experimental determination. It is found that mercury vapour, helium, argon and its associates (neon, krypton, &c.) have the value 1.67; hence we conclude that these gases exist as monatomic molecules. Oxygen, nitrogen, hydrogen and carbon monoxide have the value 1.4; these gases have diatomic molecules, a fact capable of demonstration by other means. Hence it may be inferred that this value is typical for diatomic molecules. Similarly, greater atomic complexity is reflected in a further decrease in the ratio Cp/Cv. The following table gives a comparative view of the specific heats and the ratio for molecules of variable atomic content.
The abnormal specific heats of the halogen elements may be due to a loosening of the atoms, a preliminary to the dissociation into monatomic molecules which occurs at high temperatures. In the more complex gases the specific heat varies considerably with temperature; only in the case of monatomic gases does it remain constant. Le Chatelier (Zeit. f. phys. Chem. i. 456) has given the formula Cp = 6.5 + aT, where a is a constant depending on the complexity of the molecule, as an expression for the molecular heat at constant pressure at any temperature T (reckoned on the absolute scale). For a further discussion of the ratio of the specific heats see Molecule.
| Molecular Content. | Examples. | Cp. | Cv. | Cp/Cv. |
| Monatomic | Hg, Zn, Cd, He, Ar, &c. | 5 | 3 | 1.66 |
| Diatomic | H2, 02, N2 (0°-200°) | 6.83 | 4.83 | 1.41 |
| Cl2, Br2, I2 (0°-200°) | 8.6 | 6.6 | 1.30 | |
| HCl, HBr, HI, NO, CO | . . . | . . . | 1.41 | |
| Triatomic | H2O, H2S, N2O, CO2 | 9.2 | 7.2 | 1.28 |
| Tetratomic | As4, P4 | 13.4 | 11.4 | 1.175 |
| NH3, C2H2 | 11.6 | 9.6 | 1.21 | |
| Pentatomic | CHCl3 | 14 | 12 | 1.17 |
| Hexatomic | C2H4, C2H3Br | 16.4 | 14.4 | 1.14 |
Specific Heats of Solids.—The development of the atomic theory and the subsequent determination of atomic weights in the opening decades of the 19th century inspired A.T. Petit and P.L. Dulong to investigate relations (if any) existing between specific heats and the atomic weight. Their observations on the solid elements led to a remarkable generalization, now known as Dulong and Petit’s law. This states that “the atomic heat (the product of the atomic weight and specific heat) of all elements is a constant quantity.” The value of this constant when H = 1 is about 6.4; Dulong and Petit, using O = 1, gave the value .38, the specific heat of water being unity in both cases. This law—purely empirical in origin—was strengthened by Berzelius, who redetermined many specific heats, and applied the law to determine the true atomic weight from the equivalent weight. At the same time he perceived that specific heats varied with temperature and also with allotropes, e.g. graphite and diamond. The results of Berzelius were greatly extended by Hermann Kopp, who recognized that carbon, boron and silicon were exceptions to the law. He regarded these anomalies as solely due to the chemical nature of the elements, and ignored or regarded as insignificant such factors as the state of aggregation and change of specific heat with temperature.
The specific heats of carbon, boron and silicon subsequently formed the subject of elaborate investigations by H.F. Weber, who showed that with rise of temperature the specific (and atomic) heat increases, finally attaining a fairly constant value; diamond, graphite and the various amorphous forms of carbon having the value about 5.6 at 1000°, and silicon 5.68 at 232°; while he concluded that boron attained a constant value of 5.5. Niison and Pettersson’s observations on beryllium and germanium have shown that the atomic heats of these metals increase with rise of temperature, finally becoming constant with a value 5.6. W.A. Tilden (Phil. Trans., 1900, p. 233) investigated nickel and cobalt over a wide range of temperature (from -182.5° to 100°); his results are:—
| Cobalt. | Nickel. | |
| From -182.5° to -78.4° | 4.1687 | 4.1874 |
| -78.4° to 15° | 5.4978 | 5.6784 |
| 15° to 100° | 6.0324 | 6.3143 |
It is evident that the atomic heats of these intimately associated elements approach nearer and nearer as we descend in temperature, approximating to the value 4. Other metals were tested in order to determine if their atomic heats approximated to this value at low temperatures, but with negative results.
It is apparent that the law of Dulong and Petit is not rigorously true, and that deviations are observed which invalidate the law as originally framed. Since the atomic heat of the same element varies with its state of aggregation, it must be concluded that some factor taking this into account must be introduced; moreover, the variation of specific heat with temperature introduces another factor.
We now proceed to discuss molecular heats of compounds, that is, the product of the molecular weight into the specific heat. The earliest generalization in this direction is associated with F.E. Neumann, who, in 1831, deduced from observations on many carbonates (calcium, magnesium, ferrous, zinc, barium and lead) that stoichiometric quantities (equimolecular weights) of compounds possess the same heat capacity. This is spoken of as “Neumann’s law.” Regnault confirmed Neumann’s observations, and showed that the molecular heat depended on the number of atoms present, equiatomic compounds having the same molecular heat. Kopp systematized the earlier observations, and, having made many others, he was able to show that the molecular heat was an additive property, i.e. each element retains the same heat capacity when in combination as in the free state. This has received confirmation by the researches of W.A. Tilden (Phil. Trans., 1904, 203 A, p.139) for those elements whose atomic heats vary considerably with temperature.
The specific heat of a compound may, in general, be calculated from the specific heats of its constituent elements. Conversely, if the specific heats of a compound and its constituent elements, except one, be known, then the unknown atomic heat is readily deducible. Similarly, by taking the difference of the molecular heats of compounds differing by one constituent, the molecular (or atomic) heat of this constituent is directly obtained. By this method it is shown that water, when present as “water of crystallization,” behaves as if it were ice.
Deductions from Dulong and Petit’s Law.—Denoting the atomic weight by W and the specific heat by s, Dulong and Petit’s law states that 6.4 = Ws. Thus if s be known, an approximate value of W is determinate. In the determination of the atomic weight of an element two factors must be considered: (1) its equivalent weight, i.e. the amount which is equivalent to one part of hydrogen; and (2) a factor which denotes the number of atoms of hydrogen which combines with or is equivalent to one atom of the particular element. This factor is termed the valency. The equivalent weight is capable of fairly ready determination, but the settlement of the second factor is somewhat more complex, and in this direction the law of atomic heats is of service. To take an example: 38 parts of indium combine with 35.4 parts of chlorine; hence, if the formula of the chloride be InCl, InCl2 or InCl3, indium has the atomic weights 38, 76 or 114. The specific heat of indium is 0.057; and the atomic heats corresponding to the atomic weights 38, 76 and 114 are 3.2, 4.3, 6.5. Dulong and Petit’s law thus points to the value 114, which is also supported by the position occupied by this element in the periodic classification. C. Winkler decided the atomic weight of germanium by similar reasoning.
Boiling-Point and Composition.—From the relation between the critical constants PkVk/Tk = (1/3.7)R or Tk/Pk = 3.7Vk/R, and since Vk is proportional to the volume at absolute zero, the ratio Tk/Pk should exhibit additive relations. This ratio, termed by Guye the critical coefficient, has the following approximate values:—
| C. | H. | Cl. | -O-. | =O. | N. | N=. | P. | Double linkage. | Triple linkage. |
| 1.35 | 0.57 | 2.66 | 0.87 | 1.27 | 1.6 | 1.86 | 3.01 | 0.88 | 1.03 |
Since at the boiling-point under atmospheric pressure liquids are in corresponding states, the additive nature of the critical coefficient should also be presented by boiling-points. It may be shown theoretically that the absolute boiling-point is proportional to the molecular volume, and, since this property is additive, the boiling-point should also be additive.
These relations have been more thoroughly tested in the case of organic compounds, and the results obtained agree in some measure with the deductions from molecular volumes. In general, isomers boil at about the same temperature, as is shown by the isomeric esters C9H18O2:—
| Methyl octoate | 192.9° | Amyl butyrate | 184.8° |
| Ethyl heptoate | 187.1° | Heptyl acetate | 191.3° |
| Propyl hexoate | 185.5° | Octyl formate | 198.1° |
| Butyl pentoate | 185.8 |
Equal increments in the molecule are associated with an equal rise in the boiling-point, but this increment varies in different homologous series. Thus in the normal fatty alcohols, acids, esters, nitriles and ketones, the increment per CH2 is 19°-21°; in the aldehydes it is 26°-27°. In the aromatic compounds there is no regularity between the increments due to the introduction of methyl groups into the benzene nucleus or side chains; the normal value of 20°-21° is exhibited, however, by pyridine and its derivatives. The substitution of a hydrogen atom by the hydroxyl group generally occasions a rise in boiling-point at about 100°. The same increase accompanies the introduction of the amino group into aromatic nuclei.
While certain additive relations hold between some homologous series, yet differences occur which must be referred to the constitution of the molecule. As a general rule, compounds formed Constitutive influences. with a great evolution of heat have high boiling-points, and vice versa. The introduction of negative groups into a molecule alters the boiling-point according to the number of negative groups already present. This is shown in the case of the chloracetic acids:
| CH3CO2H = 118° | Diff. | |
| ClCH2·CO2H = 185° | 67° | |
| Cl2CH·CO2H = 195° | 10° | |
| Cl3C·CO2H = 195° | -200° | 3° |
According to van ’t Hoff the substitution of chlorine atoms into a methyl group occasions the following increments:—
| Cl in CH3 | 66° |
| Cl in CH2Cl | 39° |
| Cl in CHCl2 | 13°. |
The introduction of chlorine, however, may involve a fall in the boiling-point, as is recorded by Henry in the case of the chlorinated acetonitriles:—
| NC·CH3. | NC·CH2Cl. | NC·CHC12. | NC·CC13. | |||
| 81° | 123° | 112° | 83° | |||
| 42° | -11° | -29° |
The replacement of one negative group by another is accompanied by a change in the boiling-point, which is independent of the compound in which the substitution is effected, and solely conditioned by the nature of the replaced and replacing groups. Thus bromine and iodine replace chlorine with increments of about 22° and 50° respectively.
A factor of considerable importance in determining boiling-points of isomers is the symmetry of the molecule. Referring to the esters C9H18O2 previously mentioned, it is seen that the highest boiling-points belong to methyl octoate and octyl formate, the least symmetrical, while the minimum belongs to amyl butyrate, the most symmetrical. The isomeric pentanes also exhibit a similar relation CH3(CH2)4CH3 = 38°, (CH3)2CHC2H5 = 30°, (CH3)4C = 9.5°. For a similar reason secondary alcohols boil at a lower temperature than the corresponding primary, the difference being about 19°. A.E. Earp (Phil. Mag., 1893 [5], 35, p. 458) has shown that, while an increase in molecular weight is generally associated with a rise in the boiling-point, yet the symmetry of the resulting molecule may exert such a lowering effect that the final result is a diminution in the boiling-point. The series H2S = -61°, CH3SH = 21°, (CH3)2S = 41° is an example; in the first case, the molecular weight is increased and the symmetry diminished, the increase of boiling-point being 82°; in the second case the molecular weight is again increased but the molecule assumes a more symmetrical configuration, hence the comparatively slight increase of 20°. A similar depression is presented by methyl alcohol (67°) and methyl ether (-23°).
Among the aromatic di-substitution derivatives the ortho compounds have the highest boiling-point, and the meta boil at a higher, or about the same temperature as the para compounds. Of the tri-derivatives the symmetrical compounds boil at the lowest temperature, the asymmetric next, and the vicinal at the highest.
An ethylenic or double carbon union in the aliphatic hydrocarbons has, apparently, the same effect on the boiling-point as two hydrogen atoms, since the compounds CnH2n+2 and CnH2n boil at about the same temperature. An acetylenic or triple linkage is associated with a rise in the boiling-point; for example, propargyl compounds boil about 19.5° higher than the corresponding propyl compound.
Certain regularities attend the corresponding property of the melting-point. A rule applicable to organic compounds, due to Adolf v. Baeyer and supported by F.S. Kipping (Jour. Chem. Soc., 1893, 63, p.465) states, that the melting-point of any odd member of a homologous series is lower than the melting-point of the even member containing one carbon atom less. This is true of the fatty acid series, and the corresponding ketones and alcohols, and also of the succinic acid series. Other regularities exist, but generally with many exceptions. It is to be noted that although the correlation of melting-point with constitution has not been developed to such an extent as the chemical significance of other physical properties, the melting-point is the most valuable test of the purity of a substance, a circumstance due in considerable measure to the fact that impurities always tend to lower the melting-point.
Heat of Combustion and Constitution.—In the article Thermochemistry a general account of heats of formation of chemical compounds is given, and it is there shown that this constant measures the stability of the compound. In organic chemistry it is more customary to deal with the “heat of combustion,” i.e. the heat evolved when an organic compound is completely burned in oxygen; the heat of formation is deduced from the fact that it is equal to the heats of formation of the products of combustion less the observed heat of combustion. The researches of Julius Thomsen and others have shown that in many cases definite conclusions regarding constitution can be drawn from quantitative measurements of the heats of combustion; and in this article a summary of the chief results will be given.
The identity of the four valencies of the carbon atom follows from the fact that the heats of combustion of methane, ethane, propane, trimethyl methane, and tetramethyl methane, have a constant difference in the order given, viz. 158.6 calories; this means that the replacement of a hydrogen atom by a methyl group is attended by a constant increase in the heat of combustion. The same difference attends the introduction of the methyl group into many classes of compounds, for example, the paraffins, olefines, acetylenes, aromatic hydrocarbons, alcohols, aldehydes, ketones and esters, while a slightly lower value (157.1) is found in the case of the halogen compounds, nitriles, amines, acids, ethers, sulphides and nitro compounds. It therefore appears that the difference between the heats of combustion of two adjacent members of a series of homologous compounds is practically a constant, and that this constant has two average values, viz. 158.6 and 157.1.
An important connexion between heats of combustion and constitution is found in the investigation of the effect of single, double and triple carbon linkages on the thermochemical constants. If twelve grammes of amorphous carbon be burnt to carbon dioxide under constant volume, the heat evolved (96.96 cal.) does not measure the entire thermal effect, but the difference between this and the heat required to break down the carbon molecule into atoms. If the number of atoms in the carbon molecule be denoted by n, and the heat required to split off each atom from the molecule by d, then the total heat required to break down a carbon molecule completely into atoms is nd. It follows that the true heat of combustion of carbon, i.e. the heat of combustion of one gramme-atom, is 96.96 + d. The value of d can be evaluated by considering the combustion of amorphous carbon to carbon monoxide and carbon dioxide. In the first case the thermal effect of 58.58 calories actually observed must be increased by 2d to allow for the heat absorbed in splitting off two gramme-atoms of carbon; in the second case the thermal effect of 96.96 must be increased by d as above. Now in both cases one gramme-molecule of oxygen is decomposed, and the two oxygen atoms thus formed are combined with two carbon valencies. It follows that the thermal effects stated above must be equal, i.e. 58.58 + 2d = 96.96 + d, and therefore d = 38.38. The absolute heat of combustion of a carbon atom is therefore 135.34 calories, and this is independent of the form of the carbon burned.
Consider now the combustion of a hydrocarbon of the general formula CnH2m. We assume that each carbon atom and each hydrogen atom contributes equally to the thermal effect. If α be the heat evolved by each carbon atom, and β that by each hydrogen atom, the thermal effect may be expressed as H = nα + 2mβ - A, where A is the heat required to break the molecule into its constituent atoms. If the hydrocarbon be saturated, i.e. only contain single carbon linkages, then the number of such linkages is 2n - m, and if the thermal effect of such a linkage be X, then the term A is obviously equal to (2n - m)X. The value of H then becomes H = nα + 2mβ - (2n - m)X or nξ + mν, where ξ and ν are constants. Let double bonds be present, in number p, and let the energy due to such a bond be Y. Then the number of single bonds is 2n - m - 2p, and the heat of combustion becomes H1 = nξ + mν + p(2X - Y). If triple bonds, q in number, occur also, and the energy of such a bond be Z, the equation for H becomes
H = nξ + mν + p(2X - Y) + q(3X - Z).
This is the general equation for calculating the heat of combustion of a hydrocarbon. It contains four independent constants; two of these may be calculated from the heats of combustion of saturated hydrocarbons, and the other two from the combustion of hydrocarbons containing double and triple linkages. By experiment it is found that the thermal effect of a double bond is much less than the effect of two single bonds, while a triple bond has a much smaller effect than three single bonds. J. Thomsen deduces the actual values of X, Y, Z to be 14.71, 13.27 and zero; the last value he considers to be in agreement with the labile equilibrium of acetylenic compounds. One of the most important applications of these values is found in the case of the constitution of benzene, where Thomsen decides in favour of the Claus formula, involving nine single carbon linkages, and rejects the Kekulé formula, which has three single and three double bonds (see section IV.).
The thermal effects of the common organic substituents have also been investigated. The thermal effect of the “alcohol” group C·OH may be determined by finding the heat of formation of the alcohol and subtracting the thermal effects of the remaining linkages in the molecule. The average value for primary alcohols is 44.67 cal., but many large differences from this value obtain in certain cases. The thermal effects increase as one passes from primary to tertiary alcohols, the values deduced from propyl and isopropyl alcohols and trimethyl carbinol being:—primary = 45.08, secondary = 50.39, tertiary = 60.98. The thermal effect of the aldehyde group has the average value 64.88 calories, i.e. considerably greater than the alcohol group. The ketone group corresponds to a thermal effect of 53.52 calories. It is remarkable that the difference in the heats of formation of ketones and the paraffin containing one carbon atom less is 67.94 calories, which is the heat of formation of carbon monoxide at constant volume. It follows therefore that two hydrocarbon radicals are bound to the carbon monoxide residue with the same strength as they combine to form a paraffin. The average value for the carboxyl group is 119.75 calories, i.e. it is equal to the sum of the thermal effects of the aldehyde and carbonyl groups.
The thermal effects of the halogens are: chlorine = l5.13 calories, bromine = 7.68; iodine = -4.25 calories. It is remarkable that the position of the halogen in the molecule has no effect on the heat of formation; for example, chlorpropylene and allylchloride, and also ethylene dichloride and ethylidene dichloride, have equal heats of formation. The thermal effect of the ether group has an average value of 34.31 calories. This value does not hold in the case of methylene oxide if we assign to it the formula H2C·O·CH2, but if the formula H2C·O·CH2 (which assumes the presence of two free valencies) be accepted, the calculated and observed heats of formation are in agreement.
The combination of nitrogen with carbon may result in the formation of nitriles, cyanides, or primary, secondary or tertiary amines. Thomsen deduced that a single bond between a carbon and a nitrogen gramme-atom corresponds to a thermal effect of 2.77 calories, a double bond to 5.44, and a treble bond to 8.31. From this he infers that cyanogen is C:N·N:C and not N∶C-C∶N, that hydrocyanic acid is HC·N, and acetonitrile CH3·C∶N. In the case of the amines he decides in favour of the formulae
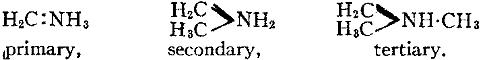
These involve pentavalent nitrogen. These formulae, however, only apply to aliphatic amines; the results obtained in the aromatic series are in accordance with the usual formulae.
Optical Relations.
Refraction and Composition.—Reference should be made to the article Refraction for the general discussion of the phenomenon known as the refraction of light. It is there shown that every substance, transparent to light, has a definite refractive index, which is the ratio of the velocity of light in vacuo to its velocity in the medium to which the refractive index refers. The refractive index of any substance varies with (1) the wave-length of the light; (2) with temperature; and (3) with the state of aggregation. The first cause of variation may be at present ignored; its significance will become apparent when we consider dispersion (vide infra).The second and third causes, however, are of greater importance, since they are associated with the molecular condition of the substance; hence, it is obvious that it is only from some function of the refractive index which is independent of temperature variations and changes of state (i.e. it must remain constant for the same substance at any temperature and in any form) that quantitative relations between refractivity and chemical composition can be derived.
The pioneer work in this field, now frequently denominated “spectro-chemistry,” was done by Sir Isaac Newton, who, from theoretical considerations based on his corpuscular theory of light, determined the function (n²-1), where n is the refractive index, to be the expression for the refractive power; dividing this expression by the density (d), he obtained (n²-1)/d, which he named the “absolute refractive power.” To P.S. Laplace is due the theoretical proof that this function is independent of temperature and pressure, and apparent experimental confirmation was provided by Biot and Arago’s, and by Dulong’s observations on gases and vapours. The theoretical basis upon which this formula was devised (the corpuscular theory) was shattered early in the 19th century, and in its place there arose the modern wave theory which theoretically invalidates Newton’s formula. The question of the dependence of refractive index on temperature was investigated in 1858 by J.H. Gladstone and the Rev. T.P. Dale; the more simple formula (n-1)/d, which remained constant for gases and vapours, but exhibited slight discrepancies when liquids were examined over a wide range of temperature, being adopted. The subject was next taken up by Hans Landolt, who, from an immense number of observations, supported in a general way the formula of Gladstone and Dale. He introduced the idea of comparing the refractivity of equimolecular quantities of different substances by multiplying the function (n-1)/d by the molecular weight (M) of the substance, and investigated the relations of chemical grouping to refractivity. Although establishing certain general relations between atomic and molecular refractions, the results were somewhat vitiated by the inadequacy of the empirical function which he employed, since it was by no means a constant which depended only on the actual composition of the substance and was independent of its physical condition. A more accurate expression (n²-1)/(n²+2)d was suggested in 1880 independently and almost simultaneously by L.V. Lorenz of Copenhagen and H.A. Lorentz of Leiden, from considerations based on the Clausius-Mossotti theory of dielectrics.
Assuming that the molecules are spherical, R.J.E. Clausius and O.F. Mossotti found a relation between the dielectric constant and the space actually occupied by the molecules, viz. K = (1 + 2a)/(1 - a), or a = (K - 1)/(K + 2), where K is the dielectric constant and a the fraction of the total volume actually occupied by matter. According to the electromagnetic theory of light K = N², where N is the refractive index for rays of infinite wave-length. Making this substitution, and dividing by d, the density of the substance, we obtain a/d = (N² - 1)/(N² + 2 )d. Since a/d is the real specific volume of the molecule, it is therefore a constant; hence (N² - 1)/(N² + 2)d is also a constant and is independent of all changes of temperature, pressure, and of the state of aggregation. To determine N recourse must be made to Cauchy’s formula of dispersion (q.v.), n = A + B/λ2 + C/λ4 + ... from which, by extrapolation, λ becoming infinite, we obtain N = A. In the case of substances possessing anomalous dispersion, the direct measurement of the refractive index for Hertzian waves of very long wave-length may be employed.
It is found experimentally that the Lorenz and Lorentz function holds fairly well, and better than the Gladstone and Dale formula. This is shown by the following observations of Rühlmann on water, the light used being the D line of the spectrum:—
| t. | (n - 1)/d. | (n² - 1)/(n² + 2)d. |
| 0 | 0.3338 | 0.2061 |
| 10 | 0.3338 | 0.2061 |
| 20 | 0.3336 | 0.2061 |
| 90 | 0.3321 | 0.2059 |
| 100 | 0.3323 | 0.2061 |
Eykmann’s observations also support the approximate constancy of the Lorenz-Lorentz formula over wide temperature differences, but in some cases the deviation exceeds the errors of observation. The values are for the Hα line:—
| Substance. | Temp. | (n² - 1)/(n² + 2)d. |
| Isosafrol, C10H10O2 | 17.6° | 0.2925 |
| 141.2° | 0.2962 | |
| Diphenyl ethylene, C14H12 | 22° | 0.3339 |
| 143.4° | 0.3382 | |
| Quinoline, C9H7N | 16.2° | 0.3187 |
| 141° | 0.3225 |
The empirical formula (n² - 1)/(n² + 0.4)d apparently gives more constant values with change of temperature than the Lorenz-Lorentz form. The superiority of the Lorenz-Lorentz formula over the Gladstone and Dale formula for changes of state is shown by the following observations of Brühl (Zeit. f. phys. Chem., 1891, 71, p. 4). The values are for the D line:—
| Substance. | Temp. | Gladstone and Dale. | Lorenz and Lorentz. | ||
| Vapour. | Liquid. | Vapour. | Liquid. | ||
| Water | 10° | 0.3101 | 0.3338 | 0.2068 | 0.2061 |
| Carbon disulphide | 10° | 0.4347 | 0.4977 | 0.2898 | 0.2805 |
| Chloroform | 10° | 0.2694 | 0.3000 | 0.1796 | 0.1790 |
Landolt and Gladstone, and at a later date J.W. Brühl, have investigated the relations existing between the refractive power and composition. To Landolt is due the proof that, Additive relations. in general, isomers, i.e. compounds having the same composition, have equal molecular refractions, and that equal differences in composition are associated with equal differences in refractive power. This is shown in the following table (the values are for Hα):—
| Substance. | Mol. Refract. | Substance. | Mol. Refract. | Diff. for CH2. | |
| Ethylene chloride | C2H4Cl2 | 20.96 | Acetic acid | 12.93 | |
| Ethylidene chloride | 21.08 | Propionic acid | 17.42 | 4.49 | |
| Fumaric acid | C4H4O4 | 70.89 | Butyric acid | 22.01 | 4.59 |
| Maleic acid | 70.29 | ||||
| o-Cresol | C7H8O | 32.52 | Acetaldehyde | 11.50 | |
| m-Cresol | 32.56 | Propionaldehyde | 15.93 | 4.43 | |
| p-Cresol | 32.57 | Butylaldehyde | 20.52 | 4.59 | |
Additive relations undoubtedly exist, but many discrepancies occur which may be assigned, as in the case of molecular volumes, to differences in constitution. Atomic refractions may be obtained either directly, by investigating the various elements, or indirectly, by considering differences in the molecular refractions of related compounds. The first method needs no explanation. The second method proceeds on the same lines as adopted for atomic volumes. By subtracting the value for CH2, which may be derived from two substances belonging to the same homologous series, from the molecular refraction of methane, CH4, the value of hydrogen is obtained; subtracting this from CH2, the value of carbon is determined. Hydroxylic oxygen is obtained by subtracting the molecular refractions of acetic acid and acetaldehyde. Similarly, by this method of differences, the atomic refraction of any element may be determined. It is found, however, that the same element has not always the same atomic refraction, the difference being due to the nature of the elements which saturate its valencies. Thus oxygen varies according as whether it is linked to hydrogen (hydroxylic oxygen), to two atoms of carbon (ether oxygen), or to one carbon atom (carbonyl oxygen); similarly, carbon varies according as whether it is singly, doubly, or trebly bound to carbon atoms.
A table of the atomic refractions and dispersions of the principal elements is here given:—
| Element. | Hα | D. | Hγ | Dispersion Hγ - Hα. |
| Hydrogen | 1.103 | 1.051 | 1.139 | 0.036 |
| Oxygen, hydroxyl | 1.506 | 1.521 | 1.525 | 0.019 |
| Oxygen, ether | 1.655 | 1.683 | 1.667 | 0.012 |
| Oxygen, carbonyl | 2.328 | 2.287 | 2.414 | 0.086 |
| Chlorine | 6.014 | 5.998 | 6.190 | 0.176 |
| Bromine | 8.863 | 8.927 | 9.211 | 0.348 |
| Iodine | 13.808 | 14.12 | 14.582 | 0.774 |
| Carbon (singly bound) | 2.365 | 2.501 | 2.404 | 0.039 |
| Double linkage of carbon | 1.836 | 1.707 | 1.859 | 0.23 |
| Triple linkage of carbon | 2.22 | 2.41 | 0.19 | |
| Nitrogen, singly bound and only to carbon | 2.76 | 2.95 | 0.19 |
Dispersion and Composition.—-In the preceding section we have seen that substances possess a definite molecular (or atomic) refraction for light of particular wave-length; the difference between the refractions for any two rays is known as the molecular (or atomic) dispersion. Since molecular refractions are independent of temperature and of the state of aggregation, it follows that molecular dispersions must be also independent of these conditions; and hence quantitative measurements should give an indication as to the chemical composition of substances. This subject has been principally investigated by Brühl; he found that molecular dispersions of liquids and gases were independent of temperature, and fairly independent of the state of aggregation, but that no simple connexion exists between atomic refractions and dispersions (see preceding table). He also showed how changes in constitution effected dispersions to a far greater extent than they did refractions; thus, while the atomic dispersion of carbon is 0.039, the dispersions due to a double and treble linkage is 0.23 and 0.19 respectively.
Colour and Constitution.—In this article a summary of the theories which have been promoted in order to connect the colour of organic compounds with their constitution will be given, and the reader is referred to the article Colour for the physical explanation of this property, and to Vision for the physiological and psychological bearings. A clear distinction must be drawn between colour and the property of dyeing; all coloured substances are not dyes, and it is shown in the article Dyeing that the property of entering into chemical or physical combination with fibres involves properties other than those essential to colour. At the same time, however, all dyestuffs are coloured substances.
A survey of coloured substances led O.N. Witt in 1876 to formulate his “chromophore-auxochrome” theory. On this theory colour is regarded as due to the presence of a “chromophore,” and dyeing power to an “auxochrome”; the latter by itself cannot produce colour or dyeing power, but it is only active in the presence of a chromophore, when it intensifies the colour and confers the property of dyeing. The principal chromophores are the azo, -N=N-, azoxy, =N2O, nitro, -NO2, nitroso, -NO, and carbonyl, =CO, groups. The azo-group is particularly active, both the aliphatic and aromatic compounds being coloured. The simplest aliphatic compounds, such as diazo-methane, diazo-ethane, and azo-formic acid, are yellow; the diamide of the latter acid is orange-red. Of the aromatic compounds azo-benzene is bright orange-red, and α-azo-naphthalene forms red needles or small steel-blue prisms. The azo-group, however, has little or no colouring effect when present in a ring system, such as in cinnolene, phthalazine and tolazone. The nitro group has a very important action mainly on account of the readiness with which it can be introduced into the molecule, but its effect is much less than that of the azo group. The colour produced is generally yellow, which, in accordance with a general rule, is intensified with an increase in the number of groups; compare, for example, mono-, di-and tri-nitrobenzene. The nitroso group is less important. The colour produced is generally of a greenish shade; for example, nitrosobenzene is green when fused or in solution (when crystalline, it is colourless), and dinitrosoresorcin has been employed as a dyestuff under the names “solid green” and “chlorine.” The carbonyl group by itself does not produce colour, but when two adjacent groups occur in the molecule, as for example in the a-diketones (such as di-acetyl and benzil), a yellow colour is produced. It also acts as a chromogenic centre when double bonds or ethylenic linkages are present, as in fluorene ketone or fluorenone.
A more complex chromophoric group is the triple ethylenic grouping
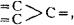 the introduction of which was rendered necessary
by the discovery of certain coloured hydrocarbons. As a general
rule, hydrocarbons are colourless; the exceptions include the golden
yellow acenaphthylene, the red bidiphenylene-ethylene, and the
derivatives of fulvene
the introduction of which was rendered necessary
by the discovery of certain coloured hydrocarbons. As a general
rule, hydrocarbons are colourless; the exceptions include the golden
yellow acenaphthylene, the red bidiphenylene-ethylene, and the
derivatives of fulvene
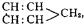 which have been discussed by J. Thiele (Ber., 1900, 33, p. 666).
This grouping is not always colour-producing, since diphenyl is colourless.
which have been discussed by J. Thiele (Ber., 1900, 33, p. 666).
This grouping is not always colour-producing, since diphenyl is colourless.
The most important auxochromes are the hydroxyl (-OH) and amino (-NH2) groups. According to the modern theory of auxochromic action, the introduction of a group into the molecule is accompanied by some strain, and the alteration in colour produced is connected with the magnitude of the strain. The amino group is more powerful than the hydroxyl, and the substituted amino group more powerful still; the repeated substitution of hydroxyl groups sometimes causes an intensification and sometimes a diminution of colour.
We may here notice an empirical rule formulated by Nietzski in 1879:—the simplest colouring substances are in the greenish-yellow and yellow, and with increasing molecular weight the colour passes into orange, red, violet, blue and green. This rule, however, is by no means perfect. Examination of the absorption spectra of coloured compounds shows that certain groupings displace the absorption bands in one direction, and other groupings in the other. If the bands be displaced towards the violet, involving a regression through the colours mentioned above, the group is said to be “hypsochromic”; if the reverse occurs the group is “bathochromic.” It may be generally inferred that an increase in molecular weight is accompanied by a change in colour in the direction of the violet.
Auxochromic groups generally aid one another, i.e. the tint deepens as the number of auxochromes increases. Also the relative position of the auxochrome to the chromophore influences colour, the ortho-position being generally the most powerful. Kauffmann (Ber., 1906, 39, p. 1959) attempted an evaluation of the effects of auxochromic groups by means of the magnetic optical constants. The method is based on the supposition that the magnetic rotation measures the strain produced in the molecule by an auxochrome, and he arranges the groups in the following order:—
| ·OCOCH3 | ·OCH3 | ·NHCOCH3 | ·NH2 | ·N(CH3)2 | ·N(C2H5)2 |
| -0.260 | 1.459 | 1.949 | 3.821 | 8.587 | 8.816 |
The phenomena attending the salt formation of coloured and colouring substances are important. The chromophoric groups are rarely strongly acid or basic; on the other hand, the auxochromes are strongly acid or basic and form salts very readily. Notable differences attend the neutralization of the chromophoric and auxochromic groups. With basic substances, the chromophoric combination with a colourless acid is generally attended by a deepening in colour; auxochromic combination, on the other hand, with a lessening. Examples of the first case are found among the colourless acridines and quinoxalines which give coloured salts; of the second case we may notice the colourless hydrochloride and sulphate of the deep yellow o-aminobenzophenone. With acid substances, the combination with “colourless” metals, i.e. metals producing colourless salts with acids, is attended by colour changes contrary to those given above, auxochromic combination being accompanied by a deepening, and chromophoric by a lessening of the tint.
Mention may be made of the phenomenon of halochromism, the name given to the power of colourless or faintly-coloured substances of combining with acids to form highly-coloured substances without the necessary production of a chromophoric group. The researches of Adolf von Baeyer and Villiger, Kehrmann, Kauffmann and others, show that this property is possessed by very many and varied substances. In many cases it may be connected with basic oxygen, and the salt formation is assumed to involve the passage of divalent into tetravalent oxygen. It seems that intermolecular change also occurs, but further research is necessary before a sound theory can be stated.
Quinone Theory of Colour.—A theory of colour in opposition to the Witt theory was proposed by Henry Armstrong in 1888 and 1892. This assumed that all coloured substances were derivatives of ortho- or para-quinone (see Quinones), and although at the time of its promotion little practical proof was given, yet the theory found wide acceptance on account of the researches of many other chemists. It follows on this theory that all coloured substances contain either of the groupings
the former being a para-quinonoid, the latter an ortho-quinonoid. While very many coloured substances must obviously contain this grouping, yet in many cases it is necessary to assume a simple intermolecular change, while in others a more complex rearrangement of bonds is necessary. Quinone, which is light yellow in colour, is the simplest coloured substance on this theory. Hydrocarbons of similar structure have been prepared by Thiele, for example, the orange-yellow tetraphenyl-para-xylylene, which is obtained by boiling the bromide C6H4[CBr(C6H5)2]2 with benzene and molecular silver. The quinonoid structure of many coloured compounds has been proved experimentally, as, for example, by Hewitt for the benzene-azo-phenols, and Hantzsch for triaminotriphenyl methane and acridine derivatives; but, at the same time, many substances cannot be so explained. A notable example is provided by the phthaleins, which result by the condensation of phthalic anhydride with phenols. In the free state these substances are colourless, and were assumed to have the formula shown in 1. Solution in dilute alkali was supposed to be accompanied by the rupture of the lactone ring with the formation of the quinonoid salt shown in 2.
Baeyer (Ber., 1905, 38, p. 569) and Silberrad (Journ. Chem. Soc., 1906, 89, p. 1787) have disputed the correctness of this explanation, and the latter has prepared melliteins and pyromelliteins, which are highly-coloured compounds produced from mellitic and pyromellitic acids, and which cannot be formulated as quinones. Baeyer has suggested that the nine carbon atom system of xanthone may act as a chromophore. An alternative view, due to Green, is that the oxygen atom of the xanthone ring is tetravalent, a supposition which permits the formulation of these substances as ortho-quinonoids.
The theories of colour have also been investigated by Hantzsch, who first considered the nitro-phenols. On the chromophore-auxochrome theory (the nitro group being the chromophore, and the hydroxyl the auxochrome) it is necessary in order to explain the high colour of the metallic salts and the colourless alkyl and aryl derivatives to assume that the auxochromic action of the hydroxyl group is only brought strongly into evidence by salt formation. Armstrong, on the other hand, assumed an intermolecular change, thus:—
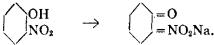
The proof of this was left for Hantzsch, who traced a connexion with the nitrolic acids of V. Meyer, which are formed when nitrous acid acts on primary aliphatic nitro compounds. Meyer formulated these compounds as nitroximes or nitro-isnitroso derivatives, viz. R·C(NO2)(NOH). Hantzsch explains the transformation of the colourless acid into red salts, which on standing yield more stable, colourless salts, by the following scheme:—
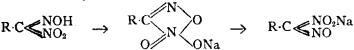
He has also shown that the nitrophenols yield, in addition to the
colourless true nitrophenol ethers, an isomeric series of coloured unstable
quinonoid aci-ethers, which have practically the same colour
and yield the same absorption spectra as the coloured metallic
salts. He suggests that the term “quinone” theory be abandoned,
and replaced by the Umlagerungs theory, since this term implies
some intermolecular rearrangement, and does not connote simply
benzenoid compounds as does “quinonoid.” H. von Liebig (Ann.,
1908, 360, p. 128), from a very complete discussion of triphenyl-methane
derivatives, concluded that the grouping
 was the only true organic chromophore, colour production, however,
requiring another condition, usually the closing of a ring.
was the only true organic chromophore, colour production, however,
requiring another condition, usually the closing of a ring.
The views as to the question of colour and constitution may be summarized as follows:—(1) The quinone theory (Armstrong, Gomberg, R. Meyer) regards all coloured substances as having a quinonoid structure. (2) The chromophore-auxochrome theory (Kauffmann) regards colour as due to the entry of an “auxochrome” into a “chromophoric” molecule. (3) If a colourless compound gives a coloured one on solution or by salt-formation, the production of colour may be explained as a particular form of ionization (Baeyer), or by a molecular rearrangement (Hantzsch). A dynamical theory due to E.C.C. Baly regards colour as due to “isorropesis” or an oscillation between the residual affinities of adjacent atoms composing the molecule.
Fluorescence and Constitution.—The physical investigation of the phenomenon named fluorescence—the property of transforming incident light into light of different refrangibility—is treated in the article Fluorescence. Researches in synthetical organic chemistry have shown that this property of fluorescence is common to an immense number of substances, and theories have been proposed whose purpose is to connect the property with constitution.
In 1897 Richard Meyer (Zeit. physik. Chemie, 24, p. 468) submitted the view that fluorescence was due to the presence of certain “fluorophore” groups; such groupings are the pyrone ring and its congeners, the central rings in anthracene and acridine derivatives, and the paradiazine ring in safranines. A novel theory, proposed by J.T. Hewitt in 1900 (Zeit. f. physik. Chemie, 34, p. 1; B.A. Report, 1903, p. 628, and later papers in the Journ. Chem. Soc.), regards the property as occasioned by internal vibrations within the molecule conditioned by a symmetrical double tautomerism, light of one wave-length being absorbed by one form, and emitted with a different wave-length by the other. This oscillation may be represented in the case of acridine and fluorescein as
This theory brings the property of fluorescence into relation with that of colour; the forms which cause fluorescence being the coloured modifications: ortho-quinonoid in the case of acridine, para-quinonoid in the case of fluorescein. H. Kauffmann (Ber., 1900, 33, p. 1731; 1904, 35, p. 294; 1905, 38, p. 789; Ann., 1906, 344, p. 30) suggested that the property is due to the presence of at least two groups. The first group, named the “luminophore,” is such that when excited by suitable aetherial vibrations emits radiant energy; the other, named the “fluorogen,” acts with the luminophore in some way or other to cause the fluorescence. This theory explains the fluorescence of anthranilic acid (o-aminobenzoic acid), by regarding the aniline residue as the luminophore, and the carboxyl group as the fluorogen, since, apparently, the introduction of the latter into the non-fluorescent aniline molecule involves the production of a fluorescent substance. Although the theories of Meyer and Hewitt do not explain (in their present form) the behaviour of anthranilic acid, yet Hewitt has shown that his theory goes far to explain the fluorescence of substances in which a double symmetrical tautomerism is possible. This tautomerism may be of a twofold nature:—(1) it may involve the mere oscillation of linkages, as in acridine; or (2) it may involve the oscillation of atoms, as in fluorescein. A theory of a physical nature, based primarily upon Sir J.J. Thomson’s theory of corpuscles, has been proposed by J. de Kowalski (Compt. rend. 1907, 144, p. 266). We may notice that ethyl oxalosuccinonitrile is the first case of a fluorescent aliphatic compound (see W. Wislicenus and P. Berg, Ber., 1908, 41, p. 3757).
Capillarity and Surface Tension.—Reference should be made to the article Capillary Action for the general discussion of this phenomenon of liquids. It is there shown that the surface tension of a liquid may be calculated from its rise in a capillary tube by the formula γ = ½rhs, where γ is the surface tension per square centimetre, r the radius of the tube, h the height of the liquid column, and s the difference between the densities of the liquid and its vapour. At the critical point liquid and vapour become identical, and, consequently, as was pointed out by Frankenheim in 1841, the surface tension is zero at the critical temperature.
Mendeléeff endeavoured to obtain a connexion between surface energy and constitution; more successful were the investigations of Schiff, who found that the “molecular surface tension,” Relation to molecular weight. which he defined as the surface tension divided by the molecular weight, is constant for isomers, and that two atoms of hydrogen were equal to one of carbon, three to one of oxygen, and seven to one of chlorine; but these ratios were by no means constant, and afforded practically no criteria as to the molecular weight of any substance.
In 1886 R. Eötvös (Wied. Ann. 27, p. 452), assuming that two liquids may be compared when the ratios of the volumes of the liquids to the volumes of the saturated vapours are the same, deduced that γV2/3 (where γ is the surface tension, and V the molecular volume of the liquid) causes all liquids to have the same temperature coefficients. This theorem was investigated by Sir W. Ramsay and J. Shields (Journ. Chem. Soc. 63, p. 1089; 65, p. 167), whose results have thrown considerable light on the subject of the molecular complexity of liquids. Ramsay and Shields suggested that there exists an equation for the surface energy of liquids, analogous to the volume-energy equation of gases, PV = RT. The relation they suspected to be of the form γS = KT, where K is a constant analogous to R, and S the surface containing one gramme-molecule, γ and T being the surface tension and temperature respectively. Obviously equimolecular surfaces are given by (Mv)2/3, where M is the molecular weight of the substance, for equimolecular volumes are Mv, and corresponding surfaces the two-thirds power of this. Hence S may be replaced by (Mv)2/3. Ramsay and Shields found from investigations of the temperature coefficient of the surface energy that T in the equation γ(Mv)2/3 = KT must be counted downwards from the critical temperature T less about 6°. Their surface energy equation therefore assumes the form γ(Mv)2/3 = K(τ - 6°). Now the value of K, γ being measured in dynes and M being the molecular weight of the substance as a gas, is in general 2.121; this value is never exceeded, but in many cases it is less. This diminution implies an association of molecules, the surface containing fewer molecules than it is supposed to. Suppose the coefficient of association be n, i.e. n is the mean number of molecules which associate to form one molecule, then by the normal equation we have γ(Mnv)2/3 = 2.121(τ - 6°); if the calculated constant be K1, then we have also γ(Mv)2/3 = K1(τ-6°). By division we obtain n2/3 = 2.121/K1, or n = (2.121/K1)3/2 the coefficient of association being thus determined.
The apparatus devised by Ramsay and Shields consisted of a capillary tube, on one end of which was blown a bulb provided with a minute hole. Attached to the bulb was a glass rod and then a tube containing iron wire. This tube was placed in an outer tube containing the liquid to be experimented with; the liquid is raised to its boiling-point, and then hermetically sealed. The whole is enclosed in a jacket connected with a boiler containing a liquid, the vapour of which serves to keep the inner tube at any desired temperature. The capillary tube can be raised or lowered at will by running a magnet outside the tube, and the heights of the columns are measured by a cathetometer or micrometer microscope.
Normal values of K were given by nitrogen peroxide, N2O4, sulphur chloride, S2Cl2, silicon tetrachloride, SiCl4, phosphorus chloride, PCl3, phosphoryl chloride, POCl3, nickel carbonyl, Ni(CO)4, carbon disulphide, benzene, pyridine, ether, methyl propyl ketone; association characterized many hydroxylic compounds: for ethyl alcohol the factor of association was 2.74-2-43, for n-propyl alcohol 2.86-2.72, acetic acid 3.62-2.77, acetone 1.26, water 3.81-2.32; phenol, nitric acid, sulphuric acid, nitroethane, and propionitril, also exhibit association.
Crystalline Form and Composition.
The development of the theory of crystal structure, and the fundamental principles on which is based the classification of crystal forms, are treated in the article Crystallography; in the same place will be found an account of the doctrine of isomorphism, polymorphism and morphotropy. Here we shall treat the latter subjects in more detail, viewed from the standpoint of the chemist. Isomorphism may be defined as the existence of two or more different substances in the same crystal form and structure, polymorphism as the existence of the same substance in two or more crystal modifications, and morphotropy (after P. von Groth) as the change in crystal form due to alterations in the molecule of closely (chemically) related substances. In order to permit a comparison of crystal forms, from which we hope to gain an insight into the prevailing molecular conditions, it is necessary that some unit of crystal dimensions must be chosen. A crystal may be regarded as built up of primitive parallelepipeda, the edges of which are in the ratio of the crystallographic axes, and the angles the axial angles of the crystals. To reduce these figures to a common standard, so that the volumes shall contain equal numbers of molecules, the notion of molecular volumes is introduced, the arbitrary values of the crystallographic axes (a, b, c) being replaced by the topic parameters18 (χ,ψ,ω), which are such that, combined with the axial angles, they enclose volumes which contain equal numbers of molecules. The actual values of the topic parameters can then readily be expressed in terms of the elements of the crystals (the axial ratios and angles), the density, and the molecular weight (see Groth, Physikalische Krystallographie, or Chemical Crystallography).
Polymorphism.—On the theory that crystal form and structure are the result of the equilibrium between the atoms and molecules composing the crystals, it is probable, a priori, that the same substance may possess different equilibrium configurations of sufficient stability, under favourable conditions, to form different crystal structures. Broadly this phenomenon is termed polymorphism; however, it is necessary to examine closely the diverse crystal modifications in order to determine whether they are really of different symmetry, or whether twinning has occasioned the apparent difference. In the article Crystallography the nature and behaviour of twinned crystals receives full treatment; here it is sufficient to say that when the planes and axes of twinning are planes and axes of symmetry, a twin would exhibit higher symmetry (but remain in the same crystal system) than the primary crystal; and, also, if a crystal approximates in its axial constants to a higher system, mimetic twinning would increase the approximation, and the crystal would be pseudo-symmetric.In general, polysymmetric and polymorphous modifications suffer transformation when submitted to variations in either temperature or pressure, or both. The criterion whether a pseudo-symmetric form is a true polymorph or not consists in the determination of the scalar properties (e.g. density, specific heat, &c.) of the original and the resulting modification, a change being in general recorded only when polymorphism exists. Change of temperature usually suffices to determine this, though in certain cases a variation in pressure is necessary; for instance, sodium magnesium uranyl acetate, NaMg(UO2)3(C2H3O2)9·9H2O shows no change in density unless the observations are conducted under a considerable pressure. Although many pseudo-symmetric twins are transformable into the simpler form, yet, in some cases, a true polymorph results, the change being indicated, as before, by alterations in scalar (as well as vector) properties.
For example, boracite forms pseudo-cubic crystals which become truly cubic at 265°, with a distinct change in density; leucite behaves similarly at about 560°. Again, the pyroxenes, RSiO3 (R = Fe, Mg, Mn, &c.), assume the forms (1) monoclinic, sometimes twinned so as to become pseudo-rhombic; (2) rhombic, resulting from the pseudo-rhombic structure of (1) becoming ultramicroscopic; and (3) triclinic, distinctly different from (1) and (2); (1) and (2) are polysymmetric modifications, while (3) and the pair (1) and (2) are polymorphs.
While polysymmetry is solely conditioned by the manner in which the mimetic twin is built up from the single crystals, there being no change in the scalar properties, and the vector properties being calculable from the nature of the twinning, in the case of polymorphism entirely different structures present themselves, both scalar and vector properties being altered; and, in the present state of our knowledge, it is impossible to foretell the characters of a polymorphous modification. We may conclude that in polymorphs the substance occurs in different phases (or molecular aggregations), and the equilibrium between these phases follows definite laws, being dependent upon temperature and pressure, and amenable to thermodynamic treatment (cf. Chemical Action and Energetics). The transformation of polymorphs presents certain analogies to the solidification of a liquid. Liquids may be cooled below their freezing-point without solidification, the metastable (after W. Ostwald) form so obtained being immediately solidified on the introduction of a particle of the solid modification; and supersaturated solutions behave in a similar manner. At the same time there may be conditions of temperature and pressure at which polymorphs may exist side by side.
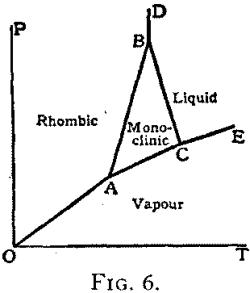
The above may be illustrated by considering the equilibrium between rhombic and monoclinic sulphur. The former, which is deposited from solutions, is transformed into monoclinic sulphur at about 96°, but with great care it is possible to overheat it and even to fuse it (at 113.5°) without effecting the transformation. Monoclinic sulphur, obtained by crystallizing fused sulphur, melts at 119.5°, and admits of undercooling even to ordinary temperatures, but contact with a fragment of the rhombic modification spontaneously brings about the transformation. From Reicher’s determinations, the exact transition point is 95.6°; it rises with increasing pressure about 0.05° for one atmosphere; the density of the rhombic form is greater than that of the monoclinic. The equilibria of these modifications may be readily represented on a pressure-temperature diagram. If OT, OP (fig. 6), be the axes of temperature and pressure, and A corresponds to the transition point (95.6°) of rhombic sulphur, we may follow out the line AB which shows the elevation of the transition point with increasing pressure. The overheating curve of rhombic sulphur extends along the curve AC, where C is the melting-point of monoclinic sulphur. The line BC, representing the equilibrium between monoclinic and liquid sulphur, is thermodynamically calculable; the point B is found to correspond to 131° and 400 atmospheres. From B the curve of equilibrium (BD) between rhombic and liquid sulphur proceeds; and from C (along CE) the curve of equilibrium between liquid sulphur and sulphur vapour. Of especial interest is the curve BD: along this line liquid and rhombic sulphur are in equilibrium, which means that at above 131° and 400 atmospheres the rhombic (and not the monoclinic) variety would separate from liquid sulphur.
Mercuric iodide also exhibits dimorphism. When precipitated from solutions it forms red tetragonal crystals, which, on careful heating, give a yellow rhombic form, also obtained by crystallization from the fused substance, or by sublimation. The transition point is 126.3° (W. Schwarz, Zeit. f. Kryst. 25, p. 613), but both modifications may exist in metastable forms at higher and lower temperatures respectively; the rhombic form may be cooled down to ordinary temperature without changing, the transformation, however, being readily induced by a trace of the red modification, or by friction. The density and specific heat of the tetragonal form are greater than those of the yellow.
Hexachlorethane is trimorphous, forming rhombic, triclinic and cubic crystals; the successive changes occur at about 44° and 71°, and are attended by a decrease in density.
Tetramorphism is exhibited by ammonium nitrate. According to O. Lehmann it melts at 168° (or at a slightly lower temperature in its water of crystallization) and on cooling forms optically isotropic crystals; at 125.6° the mass becomes doubly refracting, and from a solution rhombohedral (optically uniaxial) crystals are deposited; by further cooling acicular rhombic crystals are produced at 82.8°, and at 32.4° other rhombic forms are obtained, identical with the product obtained by crystallizing at ordinary temperatures. The reverse series of transformations occurs when this final modification is heated. M. Bellati and R. Romanese (Zeit. f. Kryst. 14, p. 78) determined the densities and specific heats of these modifications. The first and third transformations (reckoned in order with increasing temperature of the transition point) are attended by an increase in volume, the second with a contraction; the solubility follows the same direction, increasing up to 82.8°, then diminishing up to 125.6°, and then increasing from this temperature upwards.
The physical conditions under which polymorphous modifications are prepared control the form which the substance assumes. We have already seen that temperature and pressure exercise considerable influence in this direction. In the case of separation from solutions, either by crystallization or by precipitation by double decomposition, the temperature, the concentration of the solution, and the presence of other ions may modify the form obtained. In the case of sodium dihydrogen phosphate, NaH2PO4·H2O, a stable rhombic form is obtained from warm solutions, while a different, unstable, rhombic form is obtained from cold solutions. Calcium carbonate separates as hexagonal calcite from cold solutions (below 30°), and as rhombic aragonite from solutions at higher temperatures; lead and strontium carbonates, however, induce the separation of aragonite at lower temperatures. From supersaturated solutions the form unstable at the temperature of the experiment is, as a rule, separated, especially on the introduction of a crystal of the unstable form; and, in some cases, similar inoculation of the fused substance is attended by the same result. Different modifications may separate and exist side by side at one and the same time from a solution; e.g. telluric acid forms cubic and monoclinic crystals from a hot nitric acid solution, and ammonium fluosilicate gives cubic and hexagonal forms from aqueous solutions between 6° and 13°.
A comparison of the transformation of polymorphs leads to a twofold classification: (1) polymorphs directly convertible in a reversible manner—termed “enantiotropic” by O. Lehmann and (2) polymorphs in which the transformation proceeds in one direction only—termed “monotropic.” In the first class are included sulphur and ammonium nitrate; monotropy is exhibited by aragonite and calcite.
It is doubtful indeed whether any general conclusions can yet be drawn as to the relations between crystal structure and scalar properties and the relative stability of polymorphs. As a general rule the modification stable at higher temperatures possesses a lower density; but this is by no means always the case, since the converse is true for antimonious and arsenious oxides, silver iodide and some other substances. Attempts to connect a change of symmetry with stability show equally a lack of generality. It is remarkable that a great many polymorphous substances assume more symmetrical forms at higher temperatures, and a possible explanation of the increase in density of such compounds as silver iodide, &c., may be sought for in the theory that the formation of a more symmetrical configuration would involve a drawing together of the molecules, and consequently an increase in density. The insufficiency of this argument, however, is shown by the data for arsenious and antimonious oxides, and also for the polymorphs of calcium carbonate, the more symmetrical polymorphs having a lower density.
Morphotropy.—Many instances have been recorded where substitution has effected a deformation in one particular direction, the crystals of homologous compounds often exhibiting the same angles between faces situated in certain zones. The observations of Slavik (Zeit. f. Kryst., 1902, 36, p. 268) on ammonium and the quaternary ammonium iodides, of J.A. Le Bel and A. Ries (Zeit. f. Kryst., 1902, 1904, et seq.) on the substituted ammonium chlorplatinates, and of G. Mez (ibid., 1901, 35, p. 242) on substituted ureas, illustrate this point.
Ammonium iodide assumes cubic forms with perfect cubic cleavage; tetramethyl ammonium iodide is tetragonal with perfect cleavages parallel to {100} and {001}—a difference due to the lengthening of the a axes; tetraethyl ammonium iodide also assumes tetragonal forms, but does not exhibit the cleavage of the tetramethyl compound; while tetrapropyl ammonium iodide crystallizes in rhombic form. The equivalent volumes and topic parameters are tabulated:
| NH4I. | NMe4I. | NEt4I. | NPr4I. | |
| V | 57.51 | 108.70 | 162.91 | 235.95 |
| χ | 3.860 | 5.319 | 6.648 | 6.093 |
| ψ | 3.860 | 5.319 | 6.648 | 7.851 |
| ω | 3.860 | 3.842 | 3.686 | 4.933 |
From these figures it is obvious that the first three compounds form a morphotropic series; the equivalent volumes exhibit a regular progression; the values of χ and ψ, corresponding to the a axes, are regularly increased, while the value of ω, corresponding to the c axis, remains practically unchanged. This points to the conclusion that substitution has been effected in one of the cube faces. We may therefore regard the nitrogen atoms as occupying the centres of a cubic space lattice composed of iodine atoms, between which the hydrogen atoms are distributed on the tetrahedron face normals. Coplanar substitution in four hydrogen atoms would involve the pushing apart of the iodine atoms in four horizontal directions. The magnitude of this separation would obviously depend on the magnitude of the substituent group, which may be so large (in this case propyl is sufficient) as to cause unequal horizontal deformation and at the same time a change in the vertical direction.
The measure of the loss of symmetry associated with the introduction of alkyl groups depends upon the relative magnitudes of the substituent group and the rest of the molecule; and the larger the molecule, the less would be the morphotropic effect of any particular substituent. The mere retention of the same crystal form by homologous substances is not a sufficient reason for denying a morphotropic effect to the substituent group; for, in the case of certain substances crystallizing in the cubic system, although the crystal form remains unaltered, yet the structures vary. When both the crystal form and structure are retained, the substances are said to be isomorphous.
Other substituent groups exercise morphotropic effects similar to those exhibited by the alkyl radicles; investigations have been made on halogen-, hydroxy-, and nitro-derivatives of benzene and substituted benzenes. To Jaeger is due the determination of the topic parameters of certain haloid-derivatives, and, while showing that the morphotropic effects closely resemble those occasioned by methyl, he established the important fact that, in general, the crystal form depended upon the orientation of the substituents in the benzene complex.
Benzoic acid is pseudo-tetragonal, the principal axis being remarkably long; there is no cleavage at right angles to this axis. Direct nitration gives (principally) m-nitrobenzoic acid, also pseudo-tetragonal with a much shorter principal axis. From this two chlornitrobenzoic acids [COOH·NO2·Cl = 1.3.6 and 1.3.4] may be obtained. These are also pseudotetragonal; the (1.3.6) acid has nearly the same values of χ and ψ as benzoic acid, but ω is increased; compared with m-nitrobenzoic acid, χ and ψ have been diminished, whereas ω is much increased; the (1.3.4) acid is more closely related to m-nitrobenzoic acid, χ and ψ being increased, ω diminished. The results obtained for the (1.2) and (1.4) chlorbenzoic acids also illustrate the dependence of crystal form and structure on the orientation of the molecule.
The hydroxyl group also resembles the methyl group in its morphotropic effects, producing, in many cases, no change in symmetry but a dimensional increase in one direction. This holds for benzene and phenol, and is supported by the observations of Gossner on [1.3.5] trinitrobenzene and picric acid (1.3.5-trinitro, 2 oxybenzene); these last two substances assume rhombic forms, and picric acid differs from trinitrobenzene in having ω considerably greater, with χ and ψ slightly less. A similar change, in one direction only, characterizes benzoic acid and salicylic acid.
The nitro group behaves very similarly to the hydroxyl group. The effect of varying the position of the nitro group in the molecule is well marked, and conclusions may be drawn as to the orientation of the groups from a knowledge of the crystal form; a change in the symmetry of the chemical molecule being often attended by a loss in the symmetry of the crystal.
It may be generally concluded that the substitution of alkyl, nitro, hydroxyl, and haloid groups for hydrogen in a molecule occasions a deformation of crystal structure in one definite direction, hence permitting inferences as to the configuration of the atoms composing the crystal; while the nature and degree of the alteration depends (1) upon the crystal structure of the unsubstituted compound; (2) on the nature of the substituting radicle; (3) on the complexity of the substituted molecule; and (4) on the orientation of the substitution derivative.
Isomorphism.—It has been shown that certain elements and groups exercise morphotropic effects when substituted in a compound; it may happen that the effects due to two or more groups are nearly equivalent, and consequently the resulting crystal forms are nearly identical. This phenomenon was first noticed in 1822 by E. Mitscherlich, in the case of the acid phosphate and acid arsenate of potassium, KH2P(As)O4, who adopted the term isomorphism, and regarded phosphorus and arsenic as isomorphously related elements. Other isomorphously related elements and groups were soon perceived, and it has been shown that elements so related are also related chemically.
Tutton’s investigations of the morphotropic effects of the metals potassium, rubidium and caesium, in combination with the acid radicals of sulphuric and selenic acids, showed that the replacement of potassium by rubidium, and this metal in turn by caesium, was accompanied by progressive changes in both physical and crystallographical properties, such that the rubidium salt was always intermediate between the salts of potassium and caesium (see table; the space unit is taken as a pseudo-hexagonal prism). This fact finds a parallel in the atomic weights of these metals.
| V | χ | ψ | ω | |
| K2SO4 | 69.42 | 4.464 | 4.491 | 4.997 |
| Rb2SO4 | 73.36 | 4.634 | 4.664 | 5.237 |
| Cs2SO4 | 83.64 | 4.846 | 4.885 | 5.519 |
| K2SeO4 | 71.71 | 4.636 | 4.662 | 5.118 |
| Rb2SeO4 | 79.95 | 4.785 | 4.826 | 5.346 |
| Cs2SeO4 | 91.16 | 4.987 | 5.035 | 5.697 |
By taking appropriate differences the following facts will be observed: (1) the replacement of potassium by rubidium occasions an increase in the equivalent volumes by about eight units, and of rubidium by caesium by about eleven units; (2) replacement in the same order is attended by a general increase in the three topic parameters, a greater increase being met with in the replacement of rubidium by caesium; (3) the parameters χ and ψ are about equally increased, while the increase in ω is always the greatest. Now consider the effect of replacing sulphur by selenium. It will be seen that (1) the increase in equivalent volume is about 6.6; (2) all the topic parameters are increased; (3) the greatest increase is effected in the parameters χ and ψ, which are equally lengthened.
These observations admit of ready explanation in the following
manner. The ordinary structural formula of potassium sulphate is
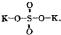 If the crystal structure be regarded as composed of
three interpenetrating point systems, one consisting of sulphur
atoms, the second of four times as many oxygen atoms, and the
third of twice as many potassium atoms, the systems being so arranged
that the sulphur system is always centrally situated with respect
to the other two, and the potassium system so that it would affect
the vertical axis, then it is obvious that the replacement of potassium
by an element of greater atomic weight would specially increase the
length of ω (corresponding to the vertical axis), and cause a smaller
increase in the horizontal parameters χ and ψ; moreover, the
increments would advance with the atomic weight of the replacing
metal. If, on the other hand, the sulphur system be replaced by a
corresponding selenium system, an element of higher atomic weight,
it would be expected that a slight increase would be observed in the
vertical parameter, and a greater increase recorded equally in the
horizontal parameters.
If the crystal structure be regarded as composed of
three interpenetrating point systems, one consisting of sulphur
atoms, the second of four times as many oxygen atoms, and the
third of twice as many potassium atoms, the systems being so arranged
that the sulphur system is always centrally situated with respect
to the other two, and the potassium system so that it would affect
the vertical axis, then it is obvious that the replacement of potassium
by an element of greater atomic weight would specially increase the
length of ω (corresponding to the vertical axis), and cause a smaller
increase in the horizontal parameters χ and ψ; moreover, the
increments would advance with the atomic weight of the replacing
metal. If, on the other hand, the sulphur system be replaced by a
corresponding selenium system, an element of higher atomic weight,
it would be expected that a slight increase would be observed in the
vertical parameter, and a greater increase recorded equally in the
horizontal parameters.
Muthmann (Zeit. f. Kryst., 1894), in his researches on the tetragonal potassium and ammonium dihydrogen phosphates and arsenates, found that the replacement of potassium by ammonium was attended by an increase of about six units in the molecular volume, and of phosphorus by arsenic by about 4.6 units. In the topic parameters the following changes were recorded: replacement of potassium by ammonium was attended by a considerable increase in ω, χ and ψ being equally, but only slightly, increased; replacement of phosphorus by arsenic was attended by a considerable increase, equally in χ and ψ, while ω suffered a smaller, but not inconsiderable, increase. It is thus seen that the ordinary plane representation of the structure of compounds possesses a higher significance than could have been suggested prior to crystallographical researches.
Identity, or approximate identity, of crystal form is not in itself sufficient to establish true isomorphism. If a substance deposits itself on the faces of a crystal of another substance of similar crystal form, the substances are probably isomorphous. Such parallel overgrowths, termed episomorphs, are very common among the potassium and sodium felspars; and K. von Hauer has investigated a number of cases in which salts exhibiting episomorphism have different colours, thereby clearly demonstrating this property of isomorphism. For example, episomorphs of white potash alum and violet chrome alum, of white magnesium sulphate and green nickel sulphate, and of many other pairs of salts, have been obtained. More useful is the property of isomorphous substances of forming mixed crystals, which are strictly isomorphous with their constituents, for all variations in composition. In such crystals each component plays its own part in determining the physical properties; in other words, any physical constant of a mixed crystal can be calculated as additively composed of the constants of the two components.
Fig. 7 represents the specific volumes of mixtures of ammonium and potassium sulphates; the ordinates representing specific volumes, and the abscissae the percentage composition of the mixture. Fig. 8 shows the variation of refractive index of mixed crystals of potash alum and thallium alum with variation in composition.
In these two instances the component crystals are miscible in all proportions; but this is by no means always the case. It may happen that the crystals do not form double salts, and are only miscible in certain proportions. Two cases then arise: (1) the properties may be expressed as linear functions of the composition, the terminal values being identical with those obtained for the individual components, and there being a break in the curve corresponding to the absence of mixed crystals; or (2) similar to (1) except that different values must be assigned to the terminal values in order to preserve collinearity. Fig. 9 illustrates the first case: the ordinates represent specific volumes, and the abscissae denote the composition of isomorphous mixtures of ammonium and potassium dihydrogen phosphates, which mutually take one another up to the extent of 20% to form homogeneous crystals. The second case is illustrated in fig. 10. Magnesium sulphate (orthorhombic) takes up ferrous sulphate (monoclinic) to the extent of 19%, forming isomorphous orthorhombic crystals; ferrous sulphate, on the other hand, takes up magnesium sulphate to the extent of 54% to form monoclinic crystals. By plotting the specific volumes of these mixed crystals as ordinates, it is found that they fall on two lines, the upper corresponding to the orthorhombic crystals, the lower to the monoclinic. From this we may conclude that these salts are isodimorphous: the upper line represents isomorphous crystals of stable orthorhombic magnesium sulphate and unstable orthorhombic ferrous sulphate, the lower line isomorphous crystals of stable monoclinic ferrous sulphate and unstable monoclinic magnesium sulphate.
An important distinction separates true mixed crystals and crystallized double salts, for in the latter the properties are not linear functions of the properties of the components; generally there is a contraction in volume, while the refractive indices and other physical properties do not, in general, obey the additive law.
Isomorphism is most clearly discerned between elements of analogous chemical properties; and from the wide generality of such observations attempts have been made to form a classification of elements based on isomorphous replacements. The following table shows where isomorphism may be generally expected. The elements are arranged in eleven series, and the series are subdivided (as indicated by semicolons) into groups; these groups exhibit partial isomorphism with the other groups of the same series (see W. Nernst, Theoretical Chemistry).
| Series | 1. Cl, Br, I, F; Mn (in permanganates). 2. S, Se; Te (in tellurides); Cr, Mn, Te (in the acids H2RO4); As, Sb (in the glances MR2). 3. As, Sb, Bi; Te (as an element); P, Vd (in salts); N, P (in organic bases). 4. K, Na, Cs, Rb, Li; Tl, Ag. 5. Ca, Ba, Sr, Pb; Fe, Zn, Mn, Mg; Ni, Co, Cu; Ce, La, Di, Er, Y, Ca; Cu, Hg, Pb; Cd, Be, In, Zn; Tl, Pb. 6. Al, Fe, Cr, Mn; Ce, U (in sesquioxides). 7. Cu, Ag (when monovalent); Au. 8. Pt, Ir, Pd, Rh, Ru, Os; Au, Fe, Ni; Sn, Te. 9. C, Si, Ti, Zr, Th, Sn; Fe, Ti. 10. Ta, Cb (Nb). 11. Mo, W, Cr. |
For a detailed comparison of the isomorphous relations of the elements the reader is referred to P. von Groth, Chemical Crystallography. Reference may also be made to Ida Freund, The Study of Chemical Composition; and to the Annual Reports of the Chemical Society for 1908, p. 258.
Bibliography.—History: F. Hoefer, Histoire de la chimie (2nd ed., 1866-1869); Hermann Kopp, Geschichte der Chemie (1869), Entwickelung der Chemie in d. neueren Zeit (1871-1874); E. von Meyer, Geschichte der Chemie (3rd ed., 1905, Eng. trans.); A. Ladenburg, Entwickelungsgeschichte der Chemie (4th ed., 1907); A. Stange, Die Zeitalter der Chemie (1908). Reference may also be made to M.M. Pattison Muir, History of Chemical Theories and Laws (1907); Ida Freund, Study of Chemical Composition (1904); T.E. Thorpe, Essays in Historical Chemistry (2nd ed., 1902). See also the article Alchemy.
Principles and Physical.—W. Ostwald, Principles of Inorganic Chemistry (3rd Eng. ed., 1908), Outlines of General Chemistry, Lehrbuch der allgemeinen Chemie; W. Nernst, Theoretische Chemie (4th ed., 1907, Eng. trans.); J.H. van’t Hoff, Lectures on Theoretical and Physical Chemistry; J. Walker, Introduction to Physical Chemistry (4th ed., 1907); H.C. Jones, Outlines of Physical Chemistry (1903); D. Mendeléeff, Principles of Chemistry (3rd ed., 1905).
Inorganic.—Roscoe and Schorlemmer, Inorganic Chemistry (3rd ed., Non-metals, 1905; Metals, 1907); R. Abegg, Handbuch der anorganischen Chemie; Gmelin-Kraut, Handbuch der anorganischen Chemie; O. Dammer, Handbuch der anorganischen Chemie; H. Moissan, Chimie minérale.
Organic.—F. Beilstein, Handbuch der organischen Chemie; M.M. Richter, Lexikon der Kohlenstoffverbindungen (these are primarily works of reference); V. Meyer and P.H. Jacobson, Lehrbuch der organischen Chemie; Richter-Anschutz, Organische Chemie (11th ed., vol. i., 1909, Eng. trans.); G.K. Schmidt, Kurzes Lehrbuch der organischen Chemie; A. Bernthsen, Organische Chemie (Eng. trans.). Practical methods are treated in Lassar-Cohn, Arbeitsmethoden für organisch-chemische Laboratorien (4th ed., 1906-1907). Select chapters are treated in A. Lachmann, Spirit of Organic Chemistry; J.B. Cohen, Organic Chemistry (1908); A.W. Stewart, Recent Advances in Organic Chemistry (1908); and in a series of pamphlets issued since 1896 with the title Sammlung chemischer und chemisch-technischer Vorträge.
Analytical.—For Blowpipe Analysis: C.F. Plattner, Probirkunst mit dem Löthrohr. For General Analysis: C.R. Fresenius, Qualitative and Quantitative Analysis, Eng. trans, by C.E. Groves (Qualitative, 1887) and A.I. Cohn (Quantitative, 1903); F.P. Treadwell, Kurzes Lehrbuch der analytischen Chemie (1905); F. Julian, Textbook of Quantitative Chemical Analysis (1904); A. Classen, Ausgewählte Methoden der analytischen Chemie (1901-1903); W. Crookes, Select Methods in Chemical Analysis (1894). Volumetric Analysis: F. Sutton, Systematic Handbook of Volumetric Analysis (1904); F. Mohr, Lehrbuch der chemisch-analytischen Titrirmethode (1896). Organic Analysis: Hans Meyer, Analyse und Konstitutionsermittlung organischer Verbindungen (1909); Wilhelm Vaubel, Die physikalischen und chemischen Methoden der quantitativen Bestimmung organischer Verbindungen. For the historical development of the proximate analysis of organic compounds see M.E.H. Dennstedt, Die Entwickelung der organischen Elementaranalyse (1899).
Encyclopaedias.—The early dictionaries of Muspratt and Watts are out of date; there is a later edition of the latter by H.F. Morley and M.M.P. Muir. A. Ladenburg, Handwörterbuch der Chemie, A. Wurtz, Dictionnaire de chimie, and F. Selmi, Enciclopedia di chimica, are more valuable; the latter two are kept up to date by annual supplements.
1 The more notable chemists of this period were Turquet de Mayerne (1573-1665), a physician of Paris, who rejected the Galenian doctrines and accepted the exaggerations of Paracelsus; Andreas Libavius (d. 1616), chiefly famous for his Opera Omnia Medicochymica (1595); Jean Baptiste van Helmont (1577-1644), celebrated for his researches on gases; F. de la Boë Sylvius (1614-1672), who regarded medicine as applied chemistry; and Otto Tachenius, who elucidated the nature of salts.
2 This dictum was questioned by the researches of H. Landolt, A. Heydweiller and others. In a series of 75 reactions it was found that in 61 there was apparently a diminution in weight, but in 1908, after a most careful repetition and making allowance for all experimental errors, Landolt concluded that no change occurred (see ELEMENT).
3 The theory of Berthollet was essentially mechanical, and he attempted to prove that the course of a reaction depended not on affinities alone but also on the masses of the reacting components. In this respect his hypothesis has much in common with the “law of mass-action” developed at a much later date by the Swedish chemists Guldberg and Waage, and the American, Willard Gibbs (see Chemical Action). In his classical thesis Berthollet vigorously attacked the results deduced by Bergman, who had followed in his table of elective attractions the path traversed by Stahl and S. F. Geoffroy.
4 Dalton’s atomic theory is treated in more detail in the article ATOM.
5 Berzelius, however, appreciated the necessity of differentiating the atom and the molecule, and even urged Dalton to amend his doctrine, but without success.
6 The following symbols were also used by Bergman:—
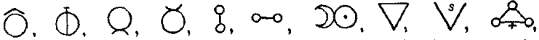
which represented zinc, manganese, cobalt, bismuth, nickel, arsenic, platinum, water, alcohol, phlogiston.
7 The following are the symbols employed by Dalton:—

which represent in order, hydrogen, nitrogen, carbon, oxygen, phosphorus, sulphur, magnesia, lime, soda, potash, strontia, baryta, mercury; iron, zinc, copper, lead, silver, platinum, and gold were represented by circles enclosing the initial letter of the element.
8 Approximate values of the atomic weights are employed here.
9 The definite distinction between potash and soda was first established by Duhamel de Monceau (1700-1781).
10 The reader is specially referred to the articles ALIZARIN; INDIGO; PURIN and TERPENES for illustrations of the manner in which chemists have artificially prepared important animal and vegetable products.
11 These observations were generalized by J.B. Dumas and Polydore Boullay (1806-1835) in their “etherin theory” (vide infra).
12 This must not be confused with the modern acetyl, CH3·CO, which at that time was known as acetoxyl.
13 It is now established that ortho compounds do exist in isomeric forms, instances being provided by chlor-, brom-, and amino-toluene, chlorphenol, and chloraniline; but arguments, e.g. E. Knoevenagel’s theory of “motoisomerism,” have been brought forward to cause these facts to support Kekulé.
14 Victor Meyer and G. Heyl (Ber., 1895, 28, p. 2776) attempted a solution from the following data. It is well known that di-ortho-substituted benzoic acids are esterified with difficulty. Two acids corresponding to the formula of Kekulé and Claus are triphenyl acrylic acid, (C6H5)2C:C(COOH)·C6H5, and triphenyl acetic acid, (C6H5)3C·COOH. Experiments showed that the second acid was much more difficult to esterify than the first, pointing to the conclusion that Claus’ formula for benzene was more probable than Kekulé’s.
15 H. Rose, Ausführliches Handbuch der analytischen Chemie (1851).
16 F. Wöhler, Die Mineralanalyse in Beispielen (1861).
17 For the connexion between valency and volume, see Valency.
18 This was done simultaneously in 1894 by W. Muthmann and A. E. H. Tutton, the latter receiving the idea from F. Becke (see Journ. Chem. Soc., 1896, 69, p. 507; 1905, 87, p. 1183).
↧ Download as ZWI file | Last modified: 11/17/2022 15:23:40 | 251 views
☰ Source: https://oldpedia.org/article/britannica11/Chemistry | License: Public domain in the USA. Project Gutenberg License
 ZWI signed:
ZWI signed: