| Congenital disorders of amino acid metabolism |
|---|
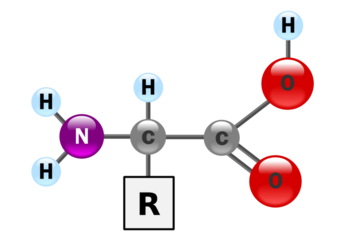 |
| The general structure of an α-amino acid, with the amino group on the left and the carboxyl group on the right. |
Inborn errors of amino acid metabolism are metabolic disorders which impair the synthesis and degradation of amino acids.
Types
- Alkaptonuria
- Aspartylglucosaminuria
- Branched-chain keto acid dehydrogenase kinase deficiency
- Methylmalonic acidemia
- Maple syrup urine disease
- Homocystinuria
- Tyrosinemia
- Trimethylaminuria
- Hartnup disease
- Biotinidase deficiency
- Ornithine carbamoyltransferase deficiency
- Carbamoyl-phosphate synthase I deficiency disease
- Citrullinemia
- Hyperargininemia
- Hyperhomocysteinemia
- Hypermethioninemia
- Hyperlysinemias
- Nonketotic hyperglycinemia
- Propionic acidemia
- Hyperprolinemia
Amino acid transport disorders
- Cystinuria
- Dicarboxylic aminoaciduria
- Hartnup disease
Amino acid storage disorders
References
External links
| Classification | D - ICD-10: E70-E72
- ICD-9-CM: 270
- MeSH: D000592
|
|---|
Inborn error of amino acid metabolism (E70–E72, 270) |
|---|
| [[Chemistry:Ketogenic amino K→acetyl-CoA]] | | Lysine/straight chain |
- Glutaric acidemia type 1
- type 2
- Hyperlysinemia
- Pipecolic acidemia
- Saccharopinuria
|
|---|
| Leucine |
- 3-hydroxy-3-methylglutaryl-CoA lyase deficiency
- 3-Methylcrotonyl-CoA carboxylase deficiency
- 3-Methylglutaconic aciduria 1
- Isovaleric acidemia
- Maple syrup urine disease
|
|---|
| Tryptophan | |
|---|
|
|---|
| G | | G→pyruvate→citrate | | Glycine |
- D-Glyceric acidemia
- Glutathione synthetase deficiency
- Sarcosinemia
- Glycine]]→Creatine: GAMT deficiency
- Glycine encephalopathy
|
|---|
|
|---|
G→glutamate→
α-ketoglutarate | | Histidine |
- Carnosinemia
- Histidinemia
- Urocanic aciduria
|
|---|
| Proline |
- Hyperprolinemia
- Prolidase deficiency
|
|---|
| Glutamate/glutamine | |
|---|
|
|---|
G→propionyl-CoA→
succinyl-CoA | | Valine |
- Hypervalinemia
- Isobutyryl-CoA dehydrogenase deficiency
- Maple syrup urine disease
|
|---|
| Isoleucine |
- 2-Methylbutyryl-CoA dehydrogenase deficiency
- Beta-ketothiolase deficiency
- Maple syrup urine disease
|
|---|
| Methionine |
- Cystathioninuria
- Homocystinuria
- Hypermethioninemia
|
|---|
| General BC/OA |
- Methylmalonic acidemia
- Methylmalonyl-CoA mutase deficiency
- Propionic acidemia
|
|---|
|
|---|
| G→fumarate | | [[Chemistry:PhenylPhenylalanine]]/tyrosine | | Phenylketonuria |
- 6-Pyruvoyltetrahydropterin synthase deficiency
- Tetrahydrobiopterin deficiency
|
|---|
| Tyrosinemia |
- Alkaptonuria/Ochronosis
- Tyrosinemia type I
- Tyrosinemia type II
- Tyrosinemia type III/Hawkinsinuria
|
|---|
| Tyrosine→Melanin |
- Albinism: Ocular albinism (1)
- Oculocutaneous albinism (Hermansky–Pudlak syndrome)
- Waardenburg syndrome
|
|---|
| Tyrosine→Norepinephrine |
- Dopamine beta hydroxylase deficiency
- reverse: Brunner syndrome
|
|---|
|
|---|
|
|---|
| G→oxaloacetate | Urea cycle/Hyperammonemia
(arginine
|
Argininemia
Argininosuccinic aciduria
Carbamoyl phosphate synthetase I deficiency
Citrullinemia
N-Acetylglutamate synthase deficiency
Ornithine transcarbamylase deficiency/translocase deficiency
|
|---|
|
|---|
|
|---|
Transport/
IE of RTT |
- Solute carrier family: Cystinuria
- Hartnup disease
- Iminoglycinuria
- Lysinuric protein intolerance
- Fanconi syndrome: Oculocerebrorenal syndrome]]
- Cystinosis
|
|---|
| Other |
- 2-Hydroxyglutaric aciduria
- Aminoacylase 1 deficiency
- Ethylmalonic encephalopathy
- Fumarase deficiency
- Trimethylaminuria
|
|---|
 | Original source: https://en.wikipedia.org/wiki/Congenital disorders of amino acid metabolism. Read more |
 From Handwiki
From Handwiki 
 KSF
KSF